2011
6
Autores:
Marcelo Rodríguez G., David Cohen S., Gabriela Egaña U., Verónica Chamy P.
Imágen 1:
Tipo de documento:
DocumentosAPOPTOSIS PLACENTARIA: MECANISMOS MOLECULARES EN LA GÉNESIS DE LA PRECLAMPSIA
Contenido del documento:
RESUMEN
Antecedentes: La placenta como otros órganos, normalmente presenta apoptosis, proceso fundamental para el mantenimiento y renovación de las estructuras. Para que este complejo proceso se realice con éxito, es necesario la estricta sincronización y modulación de las diferentes etapas de la apoptosis. Cuando la regulación falla, síndromes patológicos como la preeclampsia pueden iniciarse. Objetivo: Revisar los mecanismos moleculares implicados en la apoptosis de la placenta y sus anomalías. Método: Se realizaron búsquedas de los artículos sobre apoptosis placentaria y preeclampsia, en bases de datos Medline y otras fuentes científicas. Resultados: Muchos de los artículos confirman que la apoptosis placentaria anormal en diferentes períodos de la gestación está involucrada en la patogénesis de la preeclampsia. Conclusiones: Las anomalías de la apoptosis placentaria están relacionadas con el desarrollo posterior de preeclampsia.
PALABRAS CLAVE: Placenta, apoptosis, preeclampsia
SUMMARY
Background: Placenta like other organs, normally presents apoptosis as a fundamental process for the maintenance and renovation of structures. For this complex process to occur successfully, strict synchronization and modulation of the different stages of apoptosis are needed. When regulation of these mechanisms fails, pathologic syndromes as preeclampsia can take course. Objective: To review the molecular mechanisms involved in placental apoptosis and its anomalies. Methods: Articles about placental apoptosis and preeclampsia were searched in Medline Database and other scientific sources. Results: Many articles report and confirm that abnormal placental apoptosis at different gestational periods is involved in the pathogenesis of preeclampsia. Conclusions: Anomalies in placental apoptosis are related to the posterior development of preeclampsia.
KEY WORDS: Placenta, apoptosis, preeclampsia
INTRODUCCIÓN
El proceso de formación de la placenta incluye el desarrollo del árbol vellositario, encargado del intercambio entre el territorio materno y el territorio fetal, así como el desarrollo de porciones extravellositarias que no participan en este intercambio, pero que sí juegan un rol crucial en el proceso de placentación, mecanismo por el cual se facilita el incremento de un flujo sanguíneo adecuado a la placenta y a las porciones vellositarias de esta.
Durante este proceso existe una gran proliferación celular, que a su vez se acompaña también de la generación de una gran cantidad de desechos celulares que deben ser eliminados de manera muy eficiente, proceso conocido como apoptosis. La alteración en este proceso puede generar el desarrollo de alteraciones a nivel placentario, lo que a su vez puede generar patologías del embarazo relacionadas con un mal funcionamiento placentario.
El objetivo de este artículo es revisar los mecanismos de la apoptosis placentaria, así como sus alteraciones y su relación con entidades clínicas originadas por un desarrollo anómalo de la placenta.
TIPOS CELULARES INVOLUCRADAS EN LA PLACENTACIÓN
El trofoblasto corresponde a la capa de células que se ubican en la periferia del blastocisto y es la primera línea celular que se diferencia en el embrión, ejerciendo una acción crucial durante la implantación y placentación (1-3). Es parte de la estructura básica del árbol vellositario, conformando el epitelio del mismo y está compuesta por distintos tipos de células tales como:
1. Citotrofoblasto (CTB): corresponde a la población de “stem cell”, de la que se diferencian los otros tipos de trofoblastos.
2. Sinciciotrofoblasto (STB): responsable del intercambio de gases y nutrientes, así como también en la secreción de hormonas y péptidos.
3. Trofoblasto extravellositario (TEV): encargado de la invasión de la vasculatura endometrial y miometrial. Se distinguen 2 subtipos, TEV intersticial y TEV endovascular.
Citotrofoblasto. Corresponde a las células de Langhans. Son células mononucleadas dispuestas en las vellosidades coriónicas sobre la membrana basal del trofoblasto y recubiertas por el sinciciotrofoblasto. Corresponden a las “stem cells” pues a partir de ellas se generan los otros tipos celulares. Sus principales características son la proliferación y diferenciación celular (4).
La proliferación celular se produce a través de una multiplicación celular continua. Es capaz de ir regenerando la superficie sincicial de la vellosidad coriónica. Se ha determinado que cerca del 50% de las células del CTB se encuentran en ciclo celular (5,6). Sin embargo, existe otro sitio de proliferación trofoblástica el que corresponde a la capa celular más proximal de las vellosidades de anclaje, zona que origina al TEV.
En la diferenciación celular, el citotrofoblasto se diferencia hacia dos poblaciones celulares especializadas: STB y TEV (7). Este proceso está regulado por diferentes factores, tales como factores de transcripción, genes específicos, hormonas, factores de crecimiento, citoquinas y niveles de oxígeno (a través del HIF-1) (8,9).
Sinciciotrofoblasto: Corresponde a la monocapa epitelial que reviste la superficie vellositaria coriónica y que forma parte de la barrera placentaria que separa la sangre materna, que circula libremente en la cámara hemática de la sangre fetal. Se caracteriza por cubrir ininterrumpidamente la superficie del árbol vellositario, así como las superficies internas de la decidua basal y de la placa coriónica. Esta capa sincicial se origina por fusión de las células del CTB y está representada por citoplasma con múltiples núcleos, no observándose membranas celulares que los separen. El sinciciotrofoblasto cumple un rol fundamental en el intercambio metabólico madre-feto y viceversa, además de poseer una gran actividad endocrina, liberando distintas hormonas que participan en la homeostasis del embarazo (10,11). No posee actividad proliferativa.
Trofoblasto extravellositario: Corresponde al componente trofoblástico que se ubica fuera de las vellosidades coriónicas de intercambio. Esta población celular se genera a partir de las células del CTB de las vellosidades de anclaje (9). Luego que estas células proliferan, adquieren un fenotipo invasor por medio del cual desempeñarán su principal función: invadir el estroma uterino y luego las arterias espiraladas del endometrio, para así remodelar las paredes vasculares (12,13). De esta manera participan en la transformación de un circuito de alta resistencia al flujo sanguíneo en uno de baja resistencia (14). Se distinguen 2 subtipos de TEV (15):
TEV intersticial: invade decidua y posteriormente miometrio para luego fusionarse y convertirse en células placentarias gigantes. Incluye al TEV que no invade las paredes de los vasos.
TEV endovascular: invade las paredes de las arterias. Se distingue el trofoblasto intramural, que destruye las células musculares lisas de la capa media del vaso sanguíneo y las reemplaza por material fibrinoide y el trofoblasto intra-arterial, que forma una nueva superficie interna, reemplazando el endotelio de estos vasos maternos a través de la expresión de moléculas de adhesión que simulan un fenotipo endotelial.
De lo expuesto anteriormente se desprende que el crecimiento y mantención de la superficie epitelial que cubre el árbol vellositario depende de una proliferación celular continua del CTB (7). Por lo tanto, este trofoblasto se encuentra en permanente recambio.
RECAMBIO DEL TROFOBLASTO
El crecimiento y proliferación placentaria conlleva ramificaciónes de las vellosidades coriónicas y aumento de su superficie epitelial. Las células del STB no pueden replicarse, por lo tanto la mantención y expansión del sincicio dependerá de la incorporación continua de células desde el CTB mediante fusión sincicial. De esta única forma, el STB se mantiene funcionalmente activo (15,16).
En el otro extremo de esta remodelación continua, el material celular envejecido y desechable es vertido desde la superficie apical del STB hacia la circulación materna envueltos en una membrana protectora. El tiempo que transcurre durante este ciclo en el cual una célula del CTB es incorporada de manera transitoria dentro de este sincicio, para luego ser liberada como desecho celular en el torrente sanguíneo materno, es de aproximadamente 25 días (15,17).
MUERTE CELULAR PROGRAMADA
La muerte celular se puede clasificar en dos tipos: necrosis y muerte celular programada (18).
Necrosis. Mecanismo de lisis celular que no requiere energía para su desarrollo y en el cual hay pérdida del citoplasma y organelos hacia la matriz extracelular.
Muerte celular programada: Mecanismo que requiere energía celular para que ocurra. Se subdivide a su vez en dos tipos: autofagia y apoptosis (19). La autofagia o autoconsumo es un tipo de muerte celular descrito recientemente y que tiene fases tanto de apoptosis como de necrosis. La apoptosis se describió por primera vez en el año 1972, cuando patólogos observaron un patrón de muerte celular silenciosa, sin procesos inflamatorios, en la que los organelos experimentaban fragmentación, condensación y empaquetamiento con las membranas celulares, constituyendo estructuras densas denominadas cuerpos apoptóticos. Estos cuerpos eran posteriormente fagocitados por células vecinas o por macrófagos (20), evitando así la permanencia de restos celulares en el espacio extracelular y la respuesta inmune que estos gatillarían.
Existen signos ultraestructurales que diferencian la necrosis de la apoptosis. En esta última se observa condensación anular de la cromatina, deshidratación citoplasmática y plegamiento de la membrana, pero con conservación de organelos (21).
ACTIVACIÓN DE LA APOPTOSIS
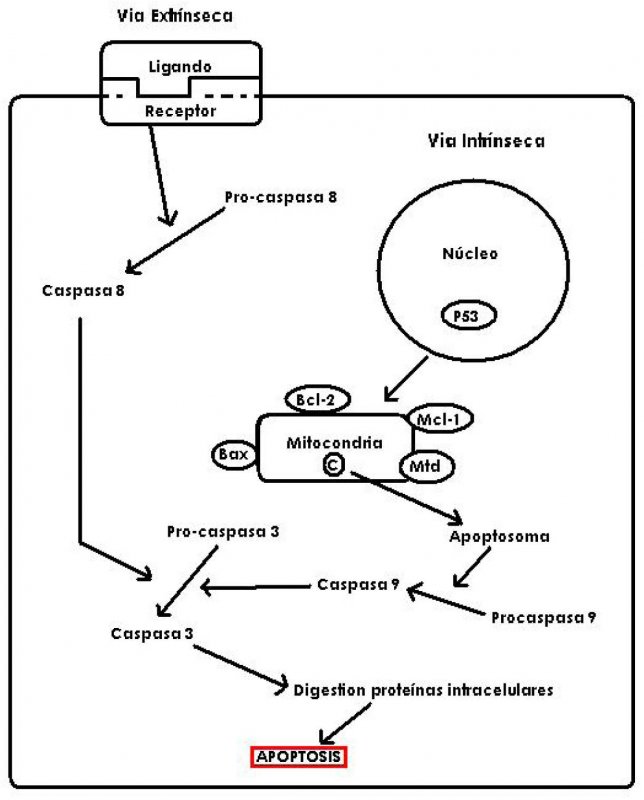
La apoptosis puede desencadenarse por una vía originada en el interior de la célula (vía intrínseca) o por una vía iniciada por una señal externa (vía extrínseca) (Figura 1). Ambas vías culminan con la activación de proteasas específicas denominadas caspasas que digieren proteínas intracelulares desencadenándose la muerte celular programada (22).
Las caspasas corresponden a una familia de 14 enzimas proteolíticas de las cuales la 2-3-6-7-8-9 y 10 están involucradas en la apoptosis (23). A su vez, se subdividen en caspasas iniciadoras del proceso de muerte celular (caspasas 2-8-9-10) y otras efectoras o ejecutoras (caspasas 3-6-7). Son sintetizadas como enzimas inactivas (procaspasas), necesitando ser divididas para adquirir su forma funcional. Una vez que son estimuladas las enzimas iniciadoras, éstas activan a las caspasas efectoras, que a su vez gatillan la acción de enzimas relacionadas con la degradación del ADN y de proteínas estructurales (24).
Vía intrínseca: puede ser programada para ocurrir en un punto específico del desarrollo celular o como respuesta a un daño celular. Esta vía se inicia con alteraciones en la permeabilidad de la membrana mitocondrial, lo que produce liberación del citocromo C. Este último se une al factor activador de proteasa apoptótica 1 (APAF-1) presente en el citoplasma, formando el apoptosoma, el cual se asocia con la pro-caspasa 9, transformándola en caspasa 9 (iniciadora), la que posteriormente activa a caspasas ejecutoras (19,25).
Vía extrínseca: utiliza un receptor que se une a un ligando pro-apoptótico. Un ejemplo es la familia de receptores del factor de necrosis tumoral (TNF), dentro de la cual destaca el TNF-R1. El TNF se une a este receptor, activando de esta manera a la caspasa 8 (26,27).
REGULACION INTRACELULAR DE LA APOPTOSIS
La regulación de la vía intrínseca de la apoptosis consta de 4 fases (19):
1. Reconocimiento de daño celular. 2. Transcripción alterada de los inhibidores de la apoptosis. 3. Alteración de la permeabilidad de la membrana mitocondrial. 4. Modificación de la actividad de las caspasas.
El reconocimiento del daño celular se realiza a través de proteínas capaces de detectar cambios en el medio ambiente intracelular, tales como HIF, DNA-PK y MAPK (28). Éstas actúan como factores de transcripción que promueven la transcripción de proteínas desencadenantes, como la p53. Esta proteína es señalada como una proteína supresora de tumores que actúa a través de la inducción de apoptosis (29).
El control de la permeabilidad de la membrana mitocondrial es llevado a cabo por la familia de proteínas Bcl-2, que contiene miembros pro-apoptóticos tales como Mtd, Bax y Bak, y también miembros antiapoptóticos como Bcl-2, Mcl-1 y Bcl-xl (30) ambos subtipos coexisten en equilibrio. La p53 modifica este balance hacia miembros proapoptóticos y de esta manera se permeabilizan poros o canales en la membrana mitocondrial, permitiendo la salida de proteínas hacia el citoplasma, tales como el citocromo C (31).
La actividad caspasa es regulada por proteínas inhibitorias de apoptosis (IAP), tales como XIAP, NIAP, survivina y livina (32).
APOPTOSIS PLACENTARIA
La apoptosis es un componente importante del desarrollo y morfogénesis embrionaria, en el cual se produce la eliminación de células únicas seleccionadas, previniendo la pérdida celular indiscriminada (33).
En este proceso, la fusión sincicial correspondería al estadío iniciador de la cascada apoptótica del CTB, mientras que la extrusión del material apoptótico desde el STB sería el resultado final del estadío efector (34,35).
Estadío iniciador: Aquí las células del CTB salen del ciclo celular, dejando de proliferar para comenzar la diferenciación y así fusionarse. Es una etapa reversible que consta de los siguientes eventos (36,37,38):
1. Unión de TNF a receptor p55 expresado en la membrana del CTB, activando y actuando sobre la procaspasa 8.
2. Caspasa 8 induce lisis de proteínas que unen el citoesqueleto a la membrana plasmática, como la alfa-fodrina; también produce externalización de fosfatidilserina (PS-flip) por fuera de la membrana plasmática, constituyéndose en la señal clave para la fusión sincicial.
3. Formación de gap junctions y secreción de moléculas fusogénicas, como conexina 43.
4. Fusión de células y formación de sincicios.
Estadío ejecutor: Es un estadío irreversible. Durante el proceso de fusión se integran los contenidos citoplasmático y nucleico del CTB hacia el STB, aportando así al STB la maquinaria completa de la cascada apoptótica.
Una vez que se ha completado la fusión sincicial, el proceso de apoptosis presenta un bloqueo inhibitorio debido a que durante la fusión también se incorporan altas concentraciones de enzimas antiapoptóticas, tales como Bcl-2 (39). De esta forma, la cascada sufre una detención, permaneciendo en un estado apoptótico latente por 3 o 4 semanas (15). Luego el proceso se reinicia con la activación de caspasa efectora 3, conduciendo a la degradación de proteínas y ácidos nucleicos.
Los eventos que ocurren en esta etapa son (15):
– Detención del proceso de apoptosis por enzimas Bcl-2, por 3-4 semanas.
– Reinicio por activación de caspasa 3.
– Activación de enzimas que degradan microfilamentos de citokeratina 18 en el citoesqueleto, lo que hace perder el anclaje del núcleo.
– Transformación y desplazamiento del núcleo, el que de forma alargada, ovoidea y rico en eucromatina, pasa después de la fusión a una forma pequeña y de cromatina densa.
– Degradación del DNA del núcleo
– Empaquetamiento de los desechos dentro de protrusiones de la membrana apical del STB, constituyéndose así los nodos sinciciales o apoptóticos
– Liberación de estos nodos apoptóticos al torrente sanguíneo materno
Los nodos apoptóticos vertidos en la sangre materna contienen núcleos sinciciales condensados, poco material citoplasmático y organelos. Estos nodos están cubiertos por membrana para evitar el contacto materno con desechos intracelulares fetales (40). De esta manera estas estructuras no inducen una gran respuesta inflamatoria materna y sólo generan una pequeña activación de células inflamatorias maternas durante el embarazo. Este material apoptótico se puede encontrar en la sangre de las venas uterinas, y de ahí se dirigen al pulmón, donde son digeridas por los macrófagos pulmonares, reduciendo sustancialmente la concentración de nodos apoptóticos en la sangre periférica materna (41,42).
APOPTOSIS Y PRECLAMPSIA
Distintos estudios evidencian que en la preeclampsia (PE), patología propia del embarazo, existiría una alteración en el proceso de diferenciación del trofoblasto vellositario por efecto del aumento de la apoptosis a este nivel (43,44,45).
Normalmente la liberación de estructuras apoptóticas desde el STB a la circulación materna no induce una respuesta inflamatoria porque los desechos celulares van cubiertos por una membrana que impide el contacto directo con estructuras maternas (33,46,47). Para que este proceso ocurra exitosamente se requiere de un período de tiempo necesario para que las distintas etapas se realicen adecuadamente, lo que normalmente toma varias semanas (40). En la preeclampsia, en cambio, hay un incremento en la proliferación y fusión del CTB, por lo que la maquinaria para el proceso de degradación subcelular se encuentra sobre-exigida. De esta manera, el mayor flujo de material celular hacia el STB, impide que la cascada apoptótica se complete en el tiempo y por tanto se verterán al torrente sanguíneo materno nodos sinciciales apoptóticamente incompletos. Esto conduce a que partes necróticas del STB desprendan y liberen componentes intracelulares parcialmente degradados y sin una total envoltura de membrana, los que en conjunto son denominados microfragmentos de sinciciotrofoblasto (STBM) (48,49).
Este desprendimiento necrótico de fragmentos intracelulares del STB, incompletamente degradados, se conoce como aponecrosis, y se refiere a la disrupción de un proceso programado dependiente de energía (apoptosis) en favor de un proceso caótico independiente de energía (necrosis), que permite el desprendimiento de contenido celular, incluidas enzimas activas, dentro de un compartimiento donde produce daño tisular (50,51).
Los estados de hipoxia placentaria se asocian con aumento de la apoptosis a nivel del trofoblasto vellositario (52,53), determinado por una menor expresión de factores antiapoptoicos como Bcl-2 (54) y por un aumento en la producción de proteínas proapoptóticas, tales como p53 (55). Esto se correlaciona con el hallazgo de que en pacientes con preeclampsia se ha documentado una mayor concentración plasmática de STBM, en comparación con las pacientes con embarazo normal (48).
Estos STBM son capaces de alterar el normal comportamiento de los macrófagos, y su aumento produce activación de neutrófilos, disrupción de células endoteliales y disminución de la relajación endotelial (56-59). De esta manera se pueden explicar tanto el daño placentario local (aumento del depósito de fibrinoide en la superficie de las vellosidades) como el daño sistémico y las manifestaciones clínicas de la preeclampsia (daño endotelial y activación del sistema inflamatorio materno).
Apoptosis en TEV: De manera similar al trofoblasto vellositario, el TEV expresa toda la maquinaria molecular de la apoptosis (15) y su regulación continúa siendo un misterio, aunque al parecer estaría influenciada por células deciduales y leucocitos maternos presentes en el lecho placentario.
TEV intersticial: Estudios iniciales señalan que en la PE existiría un aumento de la apoptosis en estas células (60). Sin embargo, estudios más recientes observan una situación distinta. Kadyrov y cols (61), reportan mediante estudios de inmunohistoquímica, que no se observan diferencias significativas en el porcentaje de apoptosis del TEV en placentas de pacientes con PE respecto a placentas de pacientes sin patología. De esto último se puede inferir que la disminución de la población celular de TEV intersticial se debería a un daño precoz en las stem cell trofoblásticas, probablemente secundario a un aumento prematuro del oxígeno a nivel local (40), por lo que no se produciría el estímulo proliferativo necesario para tener un adecuado número de células troncales
TEV endovascular: En una publicación reciente (62) se evidenció que el porcentaje de apoptosis en estas células era significativamente mayor en pacientes con PE y RCIU respecto de placentas controles, lo que era concordante con una menor densidad de trofoblasto intramural y un lúmen arterial disminuído. A juicio de los autores, lo anterior tendría relación con la mayor presencia de macrófagos inductores de la apoptosis en la vecindad de las arterias espiraladas (63-65).
Whitley y cols (66), estudiaron TEV en placentas de 12 semanas provenientes de pacientes con interrupción electiva del embarazo, y evaluaron la sensibilidad de estas células a un estímulo apoptótico determinado. A estas pacientes se les realizó previamente ecografía doppler de arterias uterinas. Las células trofoblásticas provenientes de pacientes con aumento de resistencia de las arterias uterinas mostraron una mayor sensibilidad al estímulo apoptótico, comparado con las células de TEV provenientes de pacientes con resistencia normal de arterias uterinas. De esta manera se puede plantear que el fenómeno de invasión defectuosa del trofoblasto y placentación anómala podrían estar determinados por una mayor apoptosis de estas células trofoblásticas invasoras.
CONCLUSIÓN
Las vellosidades coriónicas son la unidad anátomo-funcional de la placenta y su superficie epitelial corresponde al trofoblasto. Este último, al ser un tejido epitelial en expansión, presenta a través de un proceso de apoptosis, una renovación constante de su estructura. La regulación de este proceso a nivel placentario no está aún determinada, sin embargo, normalmente ocurre de manera sincrónica y oportuna. En algunas situaciones patológicas, la sincronía de este proceso se pierde, dando lugar a la liberación de desechos subcelulares que son capaces de activar un fenómeno de inflamación sistémica en la madre, dando pie al desarrollo de cuadros clínicos característicos.
BIBLIOGRAFÍA
1. Langman Embriología Médica, 8a ed. Sadler TW (eds.). Panamericana, 2001;132-54.
2. Norwitz E, Schust D, Fisher S. Implantation and the survival of early pregnancy. N Eng J Med 2001;345:1400-8.
3. Veenestra A, Heineman M, Faas M. The immunology of successful pregnancy. Hum Reprod Update 2003;9:347-57.
4. DeLoia JA, Burlingame JM, Karasnow JS. Differential expression of G1 cyclins during human placentogenesis. Placenta 1997;18:9-16.
5. Kosanke G, Kadyrov M, Korr H, Kaufmann P. Maternal anemia results in increased proliferation in human placental anemia results in increased proliferation in human placental villi. Trophoblas Res 1998;11:339-57.
6. Arnholdt H, Meisel F, Fandrey K, Löhrs U. Proliferation of villous trophoblast of the human placenta in normal and abnormal pregnancies. Virchows Arch B Cell Pathol Incl Mol Pathol 1991;60:365-72.
7. Goldman-Wohl D. Regulation of trophoblastic invasión: from normal implantation to pre-eclampsia. Molec Cell Endocrinol 2002;187:233-38.
8. James JA, Stone PR, Chamley LW. The regulation of throphoblast differentiation by oxygen in the first trimester of pregnancy. Human Reproduction Update 2006;12;137-44.
9. Lunghi L, Ferretti ME, Medici S, Biondi C, Vesce F. Control of human throphoblast function. Reprod Biol Endocrinol 2007;5:6. www.rbej.com./content/5/1/6
10. Huppertz B. The anatomy of the normal placenta. J Clin Pathol 2008;61:1296-302.
11. Gude N, Roberts C, Kalionis, B, King R. Growth and function of the normal human placenta. Thromb Res 2004;114:397-407.
12. Kaufmann P, Castellucci M. Extravillous trophoblast in the human placenta. A review. Trophoblast Res 1997;10:21-65.
13. Kemp B, Kertschanska S, Kadyrov M, Rath W, Kaufmann P, Huppertz B. Invasive depth of extravillous trophoblast correlates with cellular phenotype -a comparison of intra- and extrauterine implantation sites. Histochem Cell Biol 2002;117:401-14.
14. Chadda V, Viero S, Huppertz B, Kingdom J. Developmental biology of the placenta and the origins of placental insufficiency. Semin Fetal Neonat Med 2004;9:357-69.
15. Castellucci M, Kaufmann P, Baergen R. Basic structure of the villous trees. In: Bernischke K, Kaufman P (eds.), 2006. Pathology of the Human Placenta. Fifth ed. Springer, p 50-120.
16. Huppertz B, Frank HG, Reister F, Kingdom J, Korr H, Kaufmann P. Apoptosis cascade progresses during turnover of human trophoblast: Analysis of villous cytotrophoblast and syncytial fragments in vitro. Lab Invest 1999;79:1687-702.
17. Huppertz B, Frank HG, Kingdom JCP, Reister F, Kaufmann P. Villous cytotrophoblast regulation of the syncytial apoptotic cascade in the human placenta. Histochem Cell Biolm1998;110:495-508.
18. Majno G, Joris I. Apoptosis, oncosis and necrosis. An overview of cell death. Am J Pathol 1995;146:3-15.
19. Heazell AEP, Crocker IP. Live and let die: regulation of villous trophoblast apoptosis in normal pregnancies. Placenta 2008;29:772-83.
20. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972;26:239-57.
21. Kerr JFR, Gobé GC, Winterford CM, Harmon BV. Anatomical methods in cell death. Methods Cell Biol 1995;46:1-27.
22. Nunez G, Benedict MA, Hu Y, Inohara N. Caspases: the proteases of the apoptotic pathway. Oncogene 1998;17:3237-45.
23. Fraser A, Eva G. A license to kill. Cell 1996;85:781-4.
24. Huppertz B, Frank HG, Reister F, Kingdom J, Korr H, Kaufmann P. Apoptosis cascade progresses during turnover of human trophoblast: analysis of villous cytotrophoblast and syncytial fragments in vitro. Lab Invest 1999;79:1687-702.
25. Nicholson DW, Thornberry NA. Apoptosis. Life and death decisions. Science 2003; (5604):214-5.
26. Dhein J, Walczak H, Baumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature 1995;373:438-41.
27. Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature 1995;377:348-51.
28. Woo RA, McLure KG, Lees-Miller SP, Rancourt DE, Lee PW. DNA-dependent protein kinase acts upstream of p53 in response to DNA damage. Nature 1998;394:700-4.
29. Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis – the p53 network. J Cell Sci 2003;116(Pt 20):4077-85.
30. Ray J, Jurisicova A, Caniggia I. IFPA Trophoblast research award lecture: the dynamic role of Bcl-2 family members in trophoblast cell fate. Placenta 2009;23:S96-S100.
31. Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC. Channel formation by antiapoptotic protein Bcl-2. Proc Natl Acad Sci USA 1997;94:5113-8.
32. Holcik M, Gibson H, Korneluk RG. XIAP: apoptotic brake and promising herapeutic target. Apoptosis 2001;6:253-61.
33. Huppertz B, Herrler A. Regulation of proliferation and apoptosis during development of the preimplantation and the placenta. Birth Def Res C Embryo Today 2005;75:249-61.
34. Huppertz B, Frank HG, Kingdom JC, Reister F, Kaufmann P. Villous cytotrophoblast regulation of the syncytial apoptotic cascade in the human placenta. Histochem Cell Biol 1998;110:495-508.
35. Hardy K. Apoptosis in the human embryo. Rev Reprod 1999;4:125-34.
36. Yui J, Garcia-Lloret MI, Wegmann TG, Guilbert LJ. Cytotoxicity of tumor necrosis factor: alpha- and gamma-interferon against primary human placental trophoblasts. Placenta 1994;5:819-35.
37. Yui J, Hemmings D, Garcia-Lloret MI, Guilbert L. Expression of the human p55 and p75 tumor necrosis factor receptors in primary villous trophoblasts and their role in cytotoxic signal transduction. Biol Reprod 1996;55:400-9.
38. Huppertz B, Tews DS, Kaufmann P. Apoptosis and syncytial fusion in human placental trophoblast and skeletal muscle. Int Rev Cytol 2001;205:215-53.
39. Marzioni D, Mühlhauser J, Crescimanno C, Banita M, Pierleoni C, Castellucci M. BCL-2 expression in the human placenta and its correlation with fibrin deposits. Hum Reprod 1998;13:1717-22.
40. Huppertz B, Kadyrov M, Kigdom JC. Apoptosis and its role in the trophoblast. Am J Obstet Gynecol 2006;195:29-39.
41. Lunetta P, Penttila A. Immunohistochemical identification of syncytiotrophoblastic cells and megakaryocytes in pulmonary vessels in a fatal case of amniotic fluid embolism. Int J Legal Med 1996;108:210-4.
42. Johansen M, Redman CW, Wilkins T, Sargent IL. Trophoblast deportation in human pregnancy–its relevance for pre-eclampsia. Placenta 1999;20:531-9.
43. Huppertz, B, Hemmings D, Renaud S, Bulmer J, Dash P. Extravillous trophoblast apoptosis: a work shop report. Placenta 2005;26 Suppl A:S46-8.
44. Allaire AD, Ballenger KA, Wells SR, McMahon MJ, Lessey BA. Placental apoptosis in preeclampsia. Obstet Gynecol 2000;96:271-6.
45. Ishihara N, Matsuo H, Murakoshi H, Laoag-Fernandez JB, Samoto T, Maruo T. Increased apoptosis in the syncytiotrophoblast in human term placentas complicated by either preeclampsia or intrauterine growth retardation. Am J Obstet Gynecol 2002;186:158-66.
46. Mayhew TM, Leach L, McGee R, et al. Proliferation, differentiation and apoptosis in villous trophoblast at 13–41 weeks of gestation (including observations on annulate lamellae and nuclear pore complexes). Placenta 1999;20:407-22.
47. Nelson DM. Apoptotic changes occur in syncytiotrophoblast of human placental villi where fibrin type fibrinoid is deposited at discontinuities in the villous trophoblast. Placenta 1996;17:387-91.
48. Knight M, Redman CW, Linton EA, Sargent IL. Shedding of syncytiotrophoblast microvilli into the maternal circulation in pre-eclamptic pregnancies. Br J Obstet Gynaecol 1998;105:632-40.
49. Johansen M, Redman CW, Wilkins T, Sargent IL. Trophoblast deportation in human pregnancy–its relevance for pre-eclampsia. Placenta1999;20:531-9.
50. Formigli L, Papucci L, Tani A, Schiavone N, Tempestini A, Orlandini GE, et al. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J Cell Physiol 2000;182:41-9.
51. Huppertz B, Kingdom JC. Apoptosis in the trophoblastdrole of apoptosis in placental morphogenesis. J Soc Gynecol Investig 2004;11:353-62.
52. Levy R, Smith SD, Chandler K, Sadovsky Y, Nelson DM. Apoptosis in human cultured trophoblasts is enhanced by hypoxia and diminished by epidermal growth factor. Amer J Physiol Cell Physiol 2000;278:C982-8.
53. Tjoa ML, Cindrova-Davies T, Spasic-Boskovic O, Bianchi DW, Burton GJ. Trophoblastic oxidative stress and the release of cell-free feto-placental DNA. Am J Pathol 2006;169:400-4.
54. Straszewski S, Abrahams V, Mor G. The role of apoptosis in the regulation of trophoblast survival and differentiation during pregnancy. Endocrine Rev 2005;26:877-97.
55. Heazell AEP, Lacey H, Jones C, Huppertz B, et al. Effects of oxygen on cell turnover and expression of regulators of apoptosis in human placental trophoblast. Placenta 2008;29:175-86.
56. Gupta A, Hasler P, Gebhardt S, Holzgreve W, Hahn S. Occurrence of neutrophil extracellular DNA traps (NETs) in pre-eclampsia: a link with elevated levels of cell-free DNA? Ann N Y Acad Sci 2006;1075:118-22.
57. Gupta AK, Rusterholz C, Huppertz B, Malek A, Schneider H, Holzgreve W, et al. A comparative study of the effect of three different syncytiotrophoblast microparticles preparations on endothelial cells. Placenta 2005;26:59-66.
58. Hoegh AM, Tannetta D, Sargent I, Borup R, Nielsen FC, Redman C, et al. Effect of syncytiotrophoblast microvillous membrane treatment on gene expression in human umbilical vein endothelial cells. BJOG 2006;113:1270-9.
59. Cockell AP, Learmont JG, Smarason AK, Redman CW, Sargent IL, Poston L. Human placental syncytiotrophoblast microvillous membranes impair maternal vascular endothelial function. Br J Obstet Gynaecol 1997;104:235-40.
60. DiFederico E, Genbacev O, Fisher SJ. Preeclampsia is associated with widespread apoptosis of placental cytotrophoblasts within the uterine wall. Am J Pathol 1999;155:293-301.
61. Kadyrov M, Schmitz C, Black S, Kaufmann P, Huppertz B. Preeclampsia and maternal anaemia display reduced apoptosis and opposite invasive phenotypes of extravillous trophoblast. Placenta 2003;24:540-8.
62. Kadyrov M, Kingdom JCP, Huppertz B. Trophoblast invasion of spiral arteries is regulated by apoptosis: divergent features in the placental bed of pregnancies complicated by maternal anemia and early-onset preeclampsia with IUGR. Am J Obstet Gynecol 2006;194:557-63.
63. Reister F, Frank HG, Heyl W, Kosanke G, Huppertz B, Schroder W, et al. The distribution of macrophages in spiral arteries of the placental bed in pre-eclampsia differs from that in healthy patients. Placenta 1999;20:229-33.
64. Yui J, Garcia-Lloret MI, Wegmann TG, Guilbert LJ. Cytotoxicity of tumour necrosis factor-alpha and gamma-interferon against primary human placental trophoblasts. Placenta 1994;15:819-35.
65. Reister F, Frank HG, Kingdom JCP, Heyl W, Kaufmann P, Rath W. Huppertz B. Macrophage-induced apoptosis limits endovascular trophoblast invasion in the uterine wall of preeclamptic women. Lab Invest 2001;81:1143-52.
66. Whitley GS, Dash PR, Ayling LJ, Prefumo F, Thilaganathan B, Cartwright JE. Increased apoptosis in first trimester extravillous trophoblasts from pregnancies at higher risk of developing preeclampsia. Am J Pathol 2007;170:1903-9.