2012
2
Autores:
Karina Juárez R. PhD1, a, Ricardo Lara A. PhD1, b, José García O. PhD1, c
Instituciones:
1División de Genética, Centro de Investigación Biomédica de Occidente, CMNO-IMSS, Guadalajara. Doctorado en Genética Humana, Universidad de Guadalajara; Guadalajara, Jalisco, México, aBióloga., bQuímico Fármaco-Biólogo., cMédico.
Imágen 1:
Imágen 2:
Tipo de documento:
DocumentosINSUFICIENCIA OVÁRICA PREMATURA: UNA REVISIÓN
Contenido del documento:
RESUMEN
La insuficiencia ovárica prematura se caracteriza por amenorrea primaria o secundaria, hipoestrogenismo y niveles elevados de FSH (hormona folículo estimulante), que afecta a 1 de cada 100 mujeres menores de 40 años. Su etiología es multifactorial. Sólo en algunas pacientes se puede establecer una causa en particular, entre ellas: metabólicas, ambientales, autoinmunes, genéticas y iatrogénicas; siendo idiopáticos alrededor del 90% de los casos. Dentro de las causas genéticas se pueden considerar las alteraciones del cromosoma X y mutaciones en genes candidatos que regulan el desarrollo folicular. El tratamiento debe ser multidisciplinario, enfocado particularmente a la terapia de reemplazo hormonal y prevención de complicaciones. El asesoramiento genético es importante para que se tomen decisiones oportunas, sobre todo en aquellas pacientes que aún tienen la posibilidad de lograr un embarazo.
PALABRAS CLAVE: Insuficiencia ovárica prematura, genes candidatos, amenorrea
SUMMARY
Premature ovarian insufficiency is characterized by primary or secondary amenorrhea, hypoestrogenism and elevated levels of FSH (follicle stimulating hormone), which affects 1 in 100 women under 40 years. Its etiology is multifactorial, however, only in some patients can be set a particular cause, including: metabolic, environmental, autoimmune, genetic and iatrogenic, being idiopathic more than 90% of the cases. Some of the genetic causes that can be seen are the X chromosome alterations and mutations in candidate genes that regulate follicular development. Treatment should be multidisciplinary, focusing particularly on hormone replacement therapy and prevention of complications of early menopause. Genetic counseling is important for to make timely decisions, particularly in those patients who still have a chance to get a pregnancy.
KEY WORDS: Premature ovarian insufficiency, candidate genes, amenorrhea
INTRODUCCIÓN
La insuficiencia ovárica prematura (IOP) también conocida como falla ovárica prematura o menopausia prematura, se define como el cese de la ovulación y las funciones ováricas endocrinas en mujeres menores de 40 años. El término fue propuesto por primera vez en 1942 por Fuller Albright, quién es considerado el padre de la endocrinología (1,2).
Es una condición que se estima afecta a 1 de cada 100 mujeres menores de 40 años (1%), a 1 de cada 1.000 que están por debajo de los 30 años (0,1%) y a 1 de cada 10.000 mujeres con menos de 20 años de edad (0,01%) (3,4). Sin embargo, se ha descrito que de acuerdo al origen étnico los índices pueden cambiar: 1,4% para mujeres afroamericanas e hispanas, 1,0% para caucásicas, 0,5% para chinas y 0,1% para mujeres japonesas (5).
Por otra parte, se estima que para mujeres con amenorrea primaria (ausencia de la primera menarca), su prevalencia es de 10-28% y para mujeres con amenorrea secundaria (alteración en el patrón menstrual) es de 4-18% (6,7,8).
El objetivo de esta revisión es proporcionar un panorama general y actual de la IOP, que permita una mejor comprensión y manejo adecuado en la práctica clínica.
CLASIFICACIÓN
Desde el punto de vista clínico, la IOP puede dividirse en formas esporádicas (la gran mayoría de los casos) y formas familiares, donde el riesgo de recurrencia ha sido estimado hasta en un 33%, lo que confirma que la IOP es un padecimiento hereditario (9,10). En relación a los casos esporádicos, se ha reportado que alrededor del 50% de las pacientes podrían recuperar la función ovárica aún después del diagnóstico, lo que ha generado embarazos no esperados en un 5-15% (11,12).
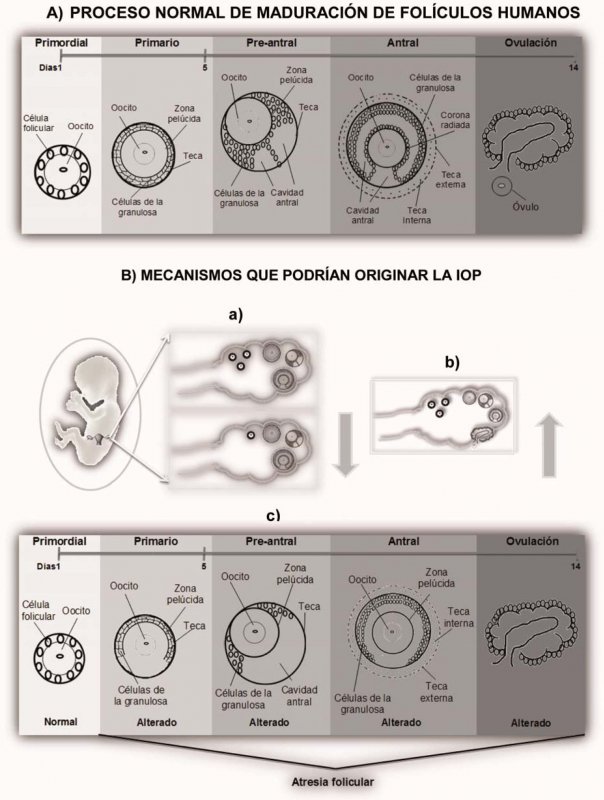
De acuerdo con la presencia o ausencia de folículos en el ovario, la IOP puede ser clasificada en dos grupos distintos histopatológicos: afolicular y folicular. En la forma afolicular, existe una depleción total de folículos ováricos y por lo tanto una pérdida permanente de la función ovárica; en cambio en la forma folicular, es posible encontrar folículos en el ovario, aunque pueden ocurrir procesos patológicos que impidan su función normal; sin embargo, en esta última condición, existe la posibilidad de regresar a una función ovárica, ya sea de forma espontánea o inducida (13,14).
DIAGNÓSTICO
En la mayoría de las mujeres, la edad de la primera menarca y ciclos menstruales son normales (15); sin embargo, la presencia de amenorrea primaria o secundaria, podrían dar la pauta para iniciar el diagnóstico de IOP (7,16). Estudios epidemiológicos han demostrado que la edad promedio en la que se diagnostica la IOP es de 33,5 ± 4,8 años, con una edad media de inicio de los síntomas de 25 años (17).
Actualmente no existen criterios diagnósticos unificados para IOP, aunque la mayoría de los clínicos consideran que este padecimiento se presenta si existe amenorrea por 3 meses o más, cuando los niveles de FSH estén por arriba de 40 mIU/ml y si los niveles de estradiol son menores de 50 pg/ml, valores que deben de ser medidos en al menos dos ocasiones en meses distintos, ya que los niveles pueden fluctuar (3,15,18).
Dentro de la historia clínica de las pacientes con IOP, es conveniente investigar antecedentes familiares, cirugías ováricas previas, radiaciones, enfermedades autoinmunes, padecimientos que involucren al cromosoma X u otras enfermedades genéticas (5). El diagnóstico de IOP puede ser confirmado con estudios de laboratorio que incluyan: ß-hCG (gonadotropina coriónica humana ß), LH (hormona luteinizante), FSH, prolactina y estradiol. Es importante realizar el cariotipo de la mujer e idealmente contar con estudios moleculares (5,15,19).
TRATAMIENTO
El mayor riesgo asociado a IOP es la deficiencia estrogénica, la cual repercute sobre la densidad mineral ósea, dando como resultado una osteoporosis consecuente, además de las enfermedades cardiovasculares y neurológicas como Alzheimer (12,20,21); sin embargo, la infertilidad sigue siendo el efecto colateral más devastador en pacientes con IOP (22).
Considerando que el diagnóstico de IOP puede afectar tanto la salud física como emocional, el tratamiento debería de ser multidisciplinario (12). Las mujeres con hipoestrogenismo requieren tomar medidas contra la osteoporosis, por lo que deben de incluir en su dieta calcio y vitamina D, sin obviar el ejercicio físico, además de evitar factores de riesgo como tabaco e ingesta de alcohol (15). Por otro lado, la terapia de reemplazo hormonal (TRH) ha disminuido los riesgos de enfermedades cardiovasculares, osteoporosis, así como el mantenimiento de la función sexual (23). Los síntomas menopáusicos tales como, bochornos, sudores nocturnos y sequedad vaginal, han sido controlados con la ingesta de píldoras anticonceptivas (15,20).
De esta forma y debido a la ausencia de tratamientos que restauren la función ovárica, una de las opciones más utilizadas para favorecer el embarazo es la donación de óvulos. Se espera que en un futuro próximo, sea posible madurar oocitos in vitro derivados de células madre, así como la preservación de tejido ovárico (20,23). En el caso de mujeres jóvenes que serán sometidas a quimioterapia o radioterapia, sería recomendable la criopreservación de oocitos (11).
Con respecto al manejo de pacientes con IOP y considerando que en las últimas décadas la tendencia a tener hijos a edades más tardías ha ido aumentado considerablemente, sería recomendable que en los casos en los que existan antecedentes familiares de IOP, se insista en el diagnóstico temprano, así como un adecuado asesoramiento genético que permita satisfacer la paridad a edades más tempranas o elegir alternativas reproductivas en clínicas de fertilidad asistida (21).
ETIOLOGÍA
Aunque la causa de IOP es desconocida en la mayoría de los casos (90%) (13), se ha propuesto que este padecimiento puede deberse a los siguientes mecanismos: a) un número inicial disminuido de folículos, b) acelerada atresia folicular ó c) que exista alteración en los procesos de maduración de los folículos (22,24) (Figura 1).
Tales mecanismos pueden ser activados por múltiples causas entre las que destacan las iatrogénicas (quimioterapia o radioterapia), metabólicas, infecciosas, toxicas, factores ambientales, autoinmunes y genéticas (4,25,26). Con respecto a la relación entre autoinmunidad e IOP, se ha estimado que del 10-30% de las pacientes adultas con este padecimiento, presentarán una enfermedad autoinmune, entre ellas hipoadrenalismo, hipotiroidismo, miastenia gravis y lupus eritematoso sistémico y aunque existen reportes que sustentan la presencia de anticuerpos anti-ováricos en suero de pacientes con IOP, la relación entre ambas aún es controversial y los mecanismos exactos son poco claros (11,15).
Ha sido ampliamente demostrada la participación genética sobre la IOP, ejemplo de ello es la monosomía del cromosoma X en el síndrome de Turner, considerada la alteración cromosómica más común asociada a IOP (27). Otras alteraciones como deleciones, translocaciones, inversiones y duplicaciones en el mismo cromosoma, han permitido identificar regiones críticas (Xp11.1 a Xp21, POF1: Xq21.3-q27 y POF2: Xq13.3-q21.1) que contienen genes importantes para la función ovárica y que han sido asociados a IOP (28,29,30).
Las deleciones terminales que ocurren en la región Xp11 pueden generar tanto casos con amenorrea primaria, como con IOP. Deleciones que involucran el brazo largo del cromosoma X generalmente resultan en IOP, aunque pueden ocurrir algunas excepciones. El fenotipo asociado a la deleción proximal Xq13 comprende amenorrea primaria, ausencia de telarca, hipogonadismo hipergonadotrófico y falla ovárica completa. Cuando la región Xq21 se encuentra involucrada en una deleción, entonces se ve comprometido el mantenimiento de la función ovárica, donde se ha estimado existen ocho diferentes genes responsables del desarrollo ovárico. Deleciones que son originadas en la región Xq25 o q26, darán como resultado amenorrea secundaria (29,31,32).
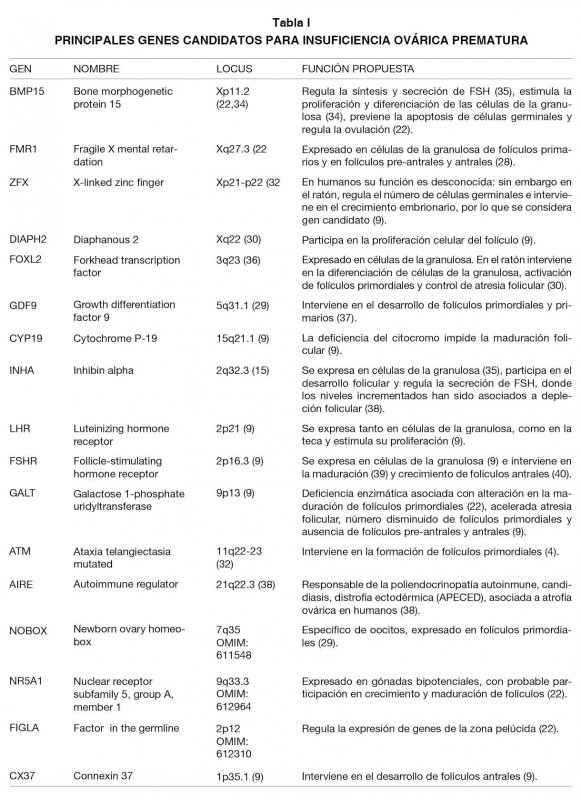
En relación a la participación de genes autosómicos, se ha descrito que la región 5q14.1-q15 contiene genes expresados en el ovario y que podrían tener participación en IOP (22); sin embargo, aunque sólo se ha asociado un pequeño número de mutaciones a IOP (FMR1, FMR2, AIRE, FSHR, LHR, GALT1, BMP15, FOXL2, EIF2B, NOGGIN, POLG e INHA), aún existen una gran cantidad de genes candidatos (DIAPH2, DFFRX, WT1, 2FX, ATM, XIST) localizados en las regiones críticas para IOP o que intervienen en alguna etapa de desarrollo del folículo, ya sea desde la formación de folículos primordiales, hasta llegar a ser folículos preovulatorios, que podrían ser analizados para dar explicación a los casos en los cuales no se ha identificado una causa en particular (idiopáticos) (8,9,29,33). En la Tabla I se presenta una lista de los genes candidatos que mayor participación tienen en la etiología de IOP.
CONCLUSIÓN
La IOP es un padecimiento heterogéneo y multifactorial, que afecta tanto la salud física como psicológica de mujeres jóvenes, por lo que es de suma importancia la colaboración de un equipo multidisciplinario que brinde una adecuada asesoría y seguimiento tanto en los casos con IOP afolicular, como en aquellos que aún tienen algo de reserva ovárica y en los cuales existe la posibilidad de lograr un embarazo.
BIBLIOGRAFÍA
1. Cooper AR, Baker VL, Sterling EW, Ryan ME, Woodruff TK, Nelson LM. The time is now for a new approach to primary ovarian insufficiency. Fertil Steril 2010;95:1890-7.
2. Pal L, Santoro N. Premature ovarian failure (POF): discordance between somatic and reproductive aging. Ageing Res Rev 2002;1:413-23.
3. Maclaran K, Horner E, Panay N. Premature ovarian failure: long-term sequelae. Menopause Int 2010;16:38-41.
4. Jagarlamudi K, Reddy P, Adhikari D, Liu K. Genetically modified mouse models for premature ovarian failure (POF). Mol Cell Endocrinol 2010;315: 1-10.
5. Nippita T, Baber R. Premature ovarian failure: a review. Climacteric 2007;10:11-22.
6. Liao C, Fu F, Yang X, Sun YM, Li DZ. Analysis of Chinese women with primary ovarian insufficiency by high resolution array-comparative genomic hybridization. Chin Med J (Engl) 2011;124:1739-42.
7. De Vos M, Devroey P, Fauser BC. Primary ovarian insufficiency. Lancet 2010; 376:911-21.
8. Suzumori N, Pangas SA, Rajkovic A. Candidate genes for premature ovarian failure. Curr Med Chem 2007;14:353-7.
9. Dixit H, Rao L, Padmalatha V, Raseswari T, Kumar A, Panda B, et al. Genes governing premature ovarian failure. Reprod Biomed Online 2010;20:724-40.
10. Ni F, Wen Q, Wang B, Zhou S, Wang J, Mu Y, Ma X, Cao Y. Mutation analysis of FOXL2 gene in Chinese patients with premature ovarian failure. Gynecol Endocrinol 2010;26:246-9.
11. Vujovic S, Brincat M, Erel T, Gambacciani M, Lambrinoudaki, Moen MH, et al. EMAS Position Statement: Managing women with premature ovarian failure. Maturitas 2010;67:91-3.
12. Kalu E, Panay N. Spontaneous premature ovarian failure: management challenges. Gynecol Endocrinol 2008;24:273-9.
13. Nelson L. Clinical practice: Primary ovarian insufficiency. N Engl J Med 2009;360:606-14.
14. Sinha P, Kuruba N. Premature ovarian failure. J Obstet Gynaecol 2007;27:16-9.
15. Shelling AN. Premature ovarian failure. Reproduction 2010;140:633-41.
16. Massin N, Czernichow C, Thibaud E, Kuttenn F, Polak M, Touraine P. Idiopathic premature ovarian failure in 63 young women. Horm Res 2006;65:89-95.
17. Coulam CB, Adamson SC, Annegers JF. Incidence of premature ovarian failure. Obstet Gynecol 67:604-10.
18. Ferreira S, Matoso E, Pinto M, Almeida J, Liehr T, Melo J, Carreira I. X-chromosome terminal deletion in a female with premature ovarian failure: Haploinsufficiency of X-linked genes as a possible explanation. Mol Cytogenet 2010;3:14.
19. Fassnacht W, Mempel A, Strowitzki T, Vogt PH. Premature ovarian failure (POF) syndrome: towards the molecular clinical analysis of its genetic complexity. Curr Med Chem 2006;13:1397-410.
20. Cartwright B, Robinson J, Rymer J. Treatment of premature ovarian failure trial: description of an ongoing clinical trial. Menopause Int 2010;16:18-22.
21. Beck-Peccoz P, Persani L. Premature ovarian failure. Orphanet J Rare Dis 2006;1:9.
22. Persani L, Rossetti R, Cacciatore C. Genes involved in human premature ovarian failure. J Mol Endocrinol 2010;45:257-79.
23. Panay N, Fenton A. Premature ovarian failure: a growing concern. Climacteric 2008;11:1-3.
24. Woad KJ, Watkins WJ, Prendergast D, Shelling AN. The genetic basis of premature ovarian failure. Aust N Z J Obstet Gynaecol 2006;46:242-4.
25. Asbagh FA, Ebrahimi M. A case report of spontaneous pregnancy during hormonal replacement therapy for premature ovarian failure. Iranian J Reprod Med 2010;9:47-9.
26. Mandon-Pépin B, Touraine P, Kuttenn F, Derbois C, Rouxel A, Matsuda F, et al. Genetic investigation of four meiotic genes in women with premature ovarian failure. Eur J Endocrinol 2008;158:107-15.
27. Cameron M, Grover S, Moore P, Jayasinghe Y. Non-chromosomal, Non-iatrogenic premature ovarian failure in an adolescent population: a case series. J Pediatr Adolesc Gynecol 2008; 21(1): 3-8.
28. Vujovic S. Aetiology of premature ovarian failure. Menopause Int 2009;15:72-5.
29. Simpson JL. Genetic and phenotypic heterogeneity in ovarian failure overview of selected candidate genes. Ann N Y Acad Sci 2008;1135:146-54.
30. Welt CK. Primary ovarian insufficiency: a more accurate term for premature ovarian failure. Clin Endocrinol (Oxf) 2008;68:499-509.
31. Simpson JL, Rajkovic A. Ovarian differentiation and gonadal failure. Am J Med Genet 1999;89:186-200.
32. Bondy C, Nelson L, Kalantaridou S. The genetic origins of ovarian failure. J Womens Health 1998;7:1225-9.
33. Ceylaner G, Altinkaya S, Mollamahmutoglu L, Ceylaner S. Genetic abnormalities in Turkish women with premature ovarian failure. Int J Gynaecol Obstet 2010;110:122-4.
34. Tiotiu B, Mercadal A, Imbert R, Verbist J, Demeestere I, De Leener A, et al. Variants of the BMP15 gene in a cohort of patients with premature ovarian failure. Hum Reprod 2010;25:1581-7.
35. Laissue P, Vinci G, Veitia R, Fellous M. Recent advances in the study of genes involved in non-syndromic premature ovarian failure. Mol Cell Endocrinol 2008;282:101-11.
36. Wang J, Liu J, Zhang Q. FOXL2 mutations in Chinese patients with blepharophimosis-ptosis-epicanthus inversus syndrome. Mol Vis 2007;13:108-13.
37. McIntosh C, Lun S, Lawrence S, Western A, McNatty K, Juengel J. The Proregion of mouse BMP15 regulates the cooperative interactions of BMP15 and GDF9. Biol Reprod 2008;79;889-96.
38. Laml T, Preyer O, Umek W, Hengstschlager M, Hanzal H. Genetic disorders in premature ovarian failure. Hum Reprod Update 2002;8:483-91.
39. Fan QR, Hendrickson WA. Structure of human follicle-stimulating hormone in complex with its receptor. Nature 2005;433:269-77.
40. Livshyts G, Podlesnaja S, Kravchenko S, Sudoma I, Livshits L. A distribution of two SNPs in exon 10 of the FSHR gene among the women with a diminished ovarian reserve in Ukraine J Assist Reprod Genet 2009;26:29-34.