2015
2
Autores:
Cristián Medina S.1, Javiera Aguirre F1, Jimena Montecinos G1, Giancarlo Schiappacasse2
Imágen 1:
Imágen 2:
Imágen 3:
Tipo de documento:
Artículo EspecialREVISIÓN PICTOGRÁFICA DE LAS ANOMALÍAS DE LOS CONDUCTOS DE MÜLLER POR RESONANCIA MAGNÉTICA
Contenido del documento:
RESUMEN
Las anomalías útero-vaginales o müllerianas son un grupo de patologías congénitas que se originan por un defecto del desarrollo, fusión o canalización de los conductos de Müller en la etapa embrionaria, excepcionalmente pueden deberse a una alteración adquirida. La mayoría se diagnostica en forma tardía, como resultado de estudios de infertilidad o complicaciones obstétricas tempranas o tardías. La resonancia magnética (RM) es un examen no invasivo, que no utiliza radiación ionizante, con una alta resolución de contraste, que permite la evaluación multiplanar de las malformaciones müllerianas, permitiendo su aproximación diagnóstica y caracterización, complementando otras técnicas de imágenes como la histerosalpingografía y la ecografía ginecológica. El radiólogo debe estar familiarizado con las alteraciones anatómicas de los genitales femeninos y su representación en RM permitiendo un diagnóstico diferencial apropiado, oportuno y en consecuencia, un tratamiento adecuado. Este artículo describe los hallazgos en este grupo de malformaciones según la clasificación de la American Fertility Society, la más utilizada en la literatura actual.
PALABRAS CLAVE: Resonancia magnética pelviana, anomalías müllerianas, malformaciones uterinas, malformaciones vaginales, infertilidad
SUMMARY
The uterus – vaginal or Müllerian anomalies are a group of congenital diseases that are caused by a developmental defect, merger or channeling of Müllerian ducts in the embryonic stage, exceptionally may be due to an acquired disorder. Most belatedly diagnosed as a result of studies of infertility or early or late obstetric complications. Magnetic resonance imaging (MRI) is a noninvasive test that does not use ionizing radiation, with high contrast resolution, which allows multiplanar evaluation of Müllerian malformations, allowing its diagnostic approach and characterization, complementing other imaging techniques such as hysterosalpingography and gynecological ultrasound.The radiologist should be familiar with the anatomical abnormalities of the female genitalia and its representation in MRI, allowing an appropriate and timely differential diagnosis, and consequently, an appropriate treatment.
KEY WORDS: Pelvic magnetic resonance, müllerian duct anomalies, uterus abnormalities, vagina abnormalities, infertility
INTRODUCCIÓN
Las malformaciones congénitas del útero, también llamadas anomalías de los conductos de Müller, derivan de un defecto en la formación, fusión o reabsorción del tabique de los conductos de Müller en los diferentes pasos del desarrollo embrionario (1), y constituyen una causa poco común de problemas ginecológicos en adolescentes y mujeres en edad fértil, manifestándose como amenorrea primaria, infertilidad o complicaciones obstétricas (1,2).
La prevalencia reportada en la población general varía de 1 a 5% y en mujeres que se realizan ecografías de causa no obstétrica es de 0,4% (3). Dichas malformaciones alcanzan una prevalencia de 8% de los casos en mujeres que consultan en centros de reproducción asistida (4,5). Se ha estimado que sólo una cuarta parte de las mujeres portadoras de estas anomalías, presentarían dificultades reproductivas (4).
La resonancia magnética (RM) provee una detallada información de la anatomía pélvica, de la morfología de los órganos genitales internos y de las malformaciones del tracto reproductivo, con una mayor sensibilidad y especificidad que la ultrasonografía y la histerosalpingografía (6,7,8).
PRESENTACIÓN CLÍNICA
El diagnóstico se suele realizar a diversas edades y en diferentes contextos clínicos, dependiendo si la anomalía es obstructiva o no obstructiva (9). En el primer caso puede presentarse de forma temprana en niñas como una masa abdomino-pélvica o perineal secundaria a la obstrucción del flujo de salida (hematocolpos), dismenorrea, sangrado vaginal anormal, amenorrea primaria y dificultad en el uso de tampones (10). Los trastornos no obstructivos se pueden diagnosticar de manera incidental, en forma posterior a la menarquia, durante el estudio de infertilidad o historia personal de abortos a repetición; en pacientes que presentan complicaciones obstétricas tardías y durante el estudio de malformaciones genitourinarias asociadas a las anomalías de los conductos de Müller (10).
EMBRIOLOGÍA
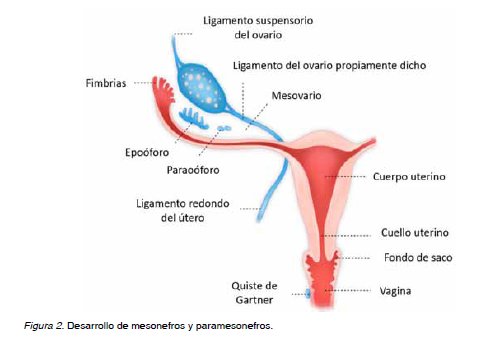
Hasta la sexta semana de vida intrauterina los embriones masculinos y femeninos no poseen diferencias, presentando ambos inicialmente los conductos paramesonéfricos (conductos de Müller) y los conductos mesonéfricos (conductos de Wolff) (11). Los primeros darán origen a la formación de las trompas de Falopio y el conducto uterino que posteriormente originaran el útero y los dos tercios superiores de la vagina (12). Los conductos mesonéfricos involucionan debido a la ausencia de expresión del gen SRY localizado en el cromosoma Y del varón, esto determina que no se sintetice el Factor Determinante Testicular, quedando como estructuras remanentes en la mujer, los ligamentos redondos, útero-ováricos y suspensorios de los ovarios (11).
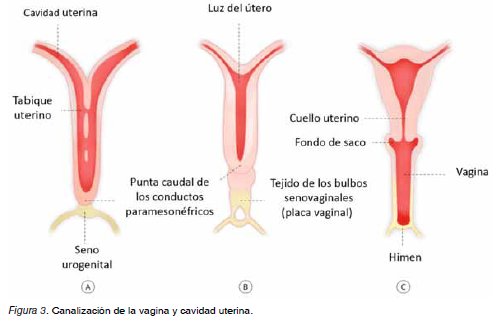
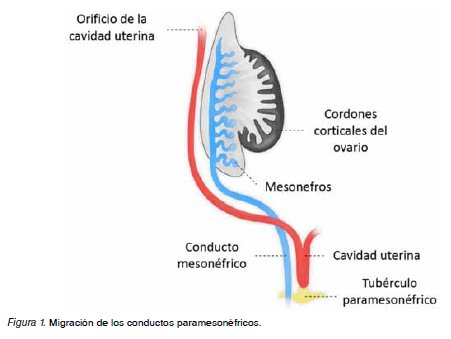
Los conductos paramesonéfricos migran caudal y medialmente, fusionando sus polos en la línea media, dando lugar en su tercio cefálico en posiciones lateralizadas y divergentes a las trompas uterinas, y en sus dos tercios distales unidos en la línea media al cuerpo uterino (Figura 1) (11). El extremo caudal de los conductos se proyecta hacia la pared posterior del seno urogenital, donde se forma el tubérculo paramesonéfrico de Müller (11,12). A partir de la 9a semana, mediante un fenómeno apoptótico mediado por el gen Bcl2, se produce una regresión del septo uterino para dar lugar a la cavidad uterina (Figura 2) (11,12).
Poco después que el extremo de los conductos paramesonéfricos alcancen el seno urogenital se generan dos evaginaciones macizas a partir de la parte pélvica del seno, llamadas bulbos seno-vaginales, que proliferan y forman la placa vaginal, que aumentará la distancia entre el útero y el seno urogenital, dando lugar a los dos tercios inferiores de la vagina, permaneciendo separados el interior de la vagina de la porción más externa del seno urogenital por el himen, que durante la vida perinatal formará un pequeño orificio (12). La canalización definitiva desde la vagina hacia el útero se produce por vacuolización de los tejidos paramesonéfricos caudal y de los bulbos seno-vaginales (Figura 3).
La formación de los ovarios depende del mesénquima y del epitelio de la cresta gonadal, sin influencia de los conductos mesonéfricos ni paramesonéfricos, no siendo afectados por las anomalías originadas en estos conductos (11,12).
Los sistemas urinario y genital derivan del mesodermo intermedio de los segmentos dorsales (6). Por lo tanto, cualquier diferenciación anormal de los conductos mesonéfricos o paramesonéfricos puede estar asociado con anomalías nefro-urológicas (10,11). La agenesia renal unilateral es la anomalía más frecuente, sin embargo también se ha descrito ectopia renal cruzada, displasia renal quística, riñón en herradura y duplicación del sistema colector (10).
TÉCNICA DE RESONANCIA MAGNÉTICA
Preparación del paciente. Las imágenes se obtienen de forma independiente del ciclo menstrual. Se utiliza gel de ultrasonido endovaginal para distender la cavidad y permitir así una mejor evaluación de la zona cervical y fondos de saco vaginales. En niñas prepuberales o con himen intacto se efectúa el examen sin gel vaginal. En aquellos casos en que las primeras imágenes muestran degradación por artefactos de peristalsis intestinal, se utiliza Buscapina 1 mg/10 kg intravenosa. Obtención de imágenes. En nuestra institución se utilizan resonadores magnéticos de 1.5 T (General Electric Medical Systems, modelo SIGNA Horizon HDxT v.23) y de 3,0 T (Siemens Medical Solutions, modelo Skyra VD11).
Se inicia la adquisición de las imágenes utilizando secuencias T2 TSE para nuestros resonadores GE 1,5 T y Siemens 3,0 T, en planos ortogonales estrictos al cuerpo uterino, en los planos axial, coronal y sagital y luego una secuencia T1 SE axial en el eje axial verdadero a la pelvis, que permite evaluar la presencia de sangre en casos de malformaciones obstructivas. En el caso que la malformación en estudio corresponda a útero bicorne, la secuencia sagital potenciada en T2 TSE deberá realizarse en forma independiente para cada cuerno, de tal manera que el lumen y eventual presencia de endometrio, se aprecie completamente en cada orientación. En el caso del útero arcuato, la secuencia axial T1 y T2, puede descomponerse en dos, una de ellas con orientación paralela al segmento anterior del arco y la segunda al segmento posterior del arco. En el caso que la malformación posea diversas angulaciones, se pueden efectuar otras secuencias T2 con angulaciones oblicuas según necesidad.
CLASIFICACIÓN DE LAS ANOMALÍAS DE LOS CONDUCTOS DE MÜLLER
La clasificación más ampliamente usada y aceptada es la realizada por la American Fertility Society (AFS) de 1988 (13,14).Esta logra una buena correlación entre las anomalías anatómicas encontradas y el momento embriológico en que se produjo la alteración de la organogénesis, clasificándose como disgenesias o trastornos de la fusión vertical o lateral (14).
Además las anomalías se clasifican como anomalías obstructivas o no obstructivas, ya que difieren en su tratamiento (9). Las anomalías obstructivas requieren atención inmediata debido al flujo retrógrado compuesto de secreciones mucosas y sangre menstrual que generan un aumento de la presión sobre los órganos circundantes, mientras que el tratamiento inmediato no se justifica en las formas no obstructivas (9).
Pese a que esta clasificación abarca la gran mayoría de las anomalías útero-vaginales, hay anomalías complejas del útero y otras dependientes del seno urogenital que quedan fuera de esta clasificación, como el septo vaginal transverso, atresia vaginal e himen imperforado (10).
En estos casos se debería limitar a describir en forma detallada y objetiva las anomalías, para no inducir a errores en los clínicos (15).
Este sistema clasifica las malformaciones en siete tipos o clases (Figura 4):
I. Hipoplasia uterina y agenesia
II. Útero unicorne
III. Útero didelfo
IV. Útero bicorne
V. Útero septado
VI. Útero arcuato
VII. Anomalías relacionadas al dietililbestrol (DES)
Clase I. Hipoplasia uterina/agenesia. Representa el 10% de las anomalías de los conductos de Müller (8). Se produce una falla temprana (alrededor de la 5ª semana de gestación) en el desarrollo embrionario, por razones desconocidas (1). La falta de desarrollo normal de los conductos de Müller causa distintos grados de agenesia o hipoplasia del útero, cérvix y dos tercios superiores de la vagina. El espectro de disgenesias incluye grados variables de alteraciones segmentarias tubáricas, del fondo y cuerpo uterino y a nivel de la vagina proximal (2,16).
El Síndrome de Mayer-Rokitansky-Kuster-Hauser (Figura 5) consiste en una anomalía combinada caracterizada por la ausencia congénita del útero, cérvix y porción superior de la vagina (1,2,13). Su incidencia es de 1/4.000-5.000 mujeres, siendo la causa más frecuente de agenesia vaginal (1/4.000- 10.000 mujeres), y la segunda causa de amenorrea primaria, sólo superada por la disgenesia gonadal (17). La función y morfología ovárica son normales, sin embargo, pueden encontrase en posiciones ectópicas fuera de la pelvis, lo que podría hacer que se reporten como ausentes, implicando consecuencias en caso de terapias de fertilidad (18,19,20).
Clase II. Útero unicorne (Figura 6). Representa el 20% de las anomalías müllerianas (21). Esta anomalía es producida por una agenesia unilateral del conducto de Müller. El útero es de pequeño tamaño, de forma curva y elongada, presenta un cuerno uterino y una trompa de Falopio lateralizados, que le dan la apariencia de un útero con “forma de plátano” (13,21). Esta anomalía describe cuatro subtipos: sin cuerno rudimentario, con cuerno rudimentario con cavidad comunicante hacia el lado normal, con cuerno rudimentario con cavidad no comunicante hacia el lado normal y útero unicorne con cuerno rudimentario sin cavidad. El útero unicorne sin cuerno rudimentario, tiene una frecuencia de 35% de los casos. Cuando está presente un cuerno rudimentario, no presenta cavidad en un 33% de los casos, siendo no comunicante en un 22% y comunicante en un 10% cuerno rudimentario sin cavidad uterina (13). La presencia de endometrio en un cuerno rudimentario no comunicante es un hallazgo importante que debe ser consignado, debido a que el flujo retrógrado puede causar endometriosis y adenomiosis, asimismo en caso de un cuerno no comunicante se puede producir hematometra e hematosalpinx (13,21).
Clase III. Útero didelfo. (Figura 7). Constituye el 5% de las anomalías müllerianas (12). Es el resultado de una falla en la fusión lateral de los conductos, desarrollándose cada conducto en forma independiente, con lo cual hay dos cavidades uterinas, que no se comunican entre sí, presencia de dos cuellos uterinos (bicollis) y 2 vaginas proximales, esta duplicación completa se presenta en dos tercios de los casos (13). Esta anomalía se asocia con un septo longitudinal vaginal en un 75% de los casos (2). En raras ocasiones se asocia a un septo vaginal transverso que causa hematometrocolpos y endometriosis en forma secundaria (1,13). Característicamente el útero didelfo presenta cuernos ampliamente divergentes (13). En ausencia de obstrucción vaginal el útero didelfo es completamente asintomático (10).
Clase IV. Útero bicorne. (Figura 8). Es el resultado de la fusión incompleta de las porciones cefálicas de los conductos müllerianos, dando cuenta de un 10% de las anomalías de los conductos de Müller (12). Consiste en dos cavidades uterinas simétricas, cada una con una cavidad endometrial, sin embargo, cada cuerno uterino no está completamente desarrollado y son de menor tamaño que en el útero didelfo. Esta anomalía está caracterizada por la presencia de una hendidura en el contorno externo del fondo uterino mayor de 1 cm, que ayuda a distinguirlo del útero septado (1,22). Las cavidades uterinas se encuentran comunicadas en su porción caudal, más frecuentemente a nivel del istmo uterino. Se clasifica en útero bicorne completo si la hendidura se extiende hasta el orificio cervical interno y en útero bicorne parcial si se encuentra confinada al fondo uterino (22).
Cuando la hendidura de un útero bicorne completo alcanza el orificio cervical interno se clasifica cono un útero bicorne con un cérvix simple (bicorne unicollis) y si la duplicación alcanza el orificio cervical externo se clasifica como útero bicorne con duplicación del cérvix (útero bicorne bicollis) (22). Si a la presencia de un útero bicorne bicollis se agrega la presencia de un septo vaginal longitudinal (25% de los casos) se hace indistinguible de un útero didelfo (13).
Clase V. Útero septado. (Figura 9). Esta malformación da cuenta del 55% de los casos de malformaciones müllerianas (23). Es resultado de un defecto de la regresión del septo útero-vaginal después de la fusión de los conductos paramesonéfricos, que puede ser completo cuando alcanza el orificio cervical externo o parcial en caso de terminar en posición más cefálica. El útero septado presenta uno de los peores resultados reproductivos, con una alta tasa de abortos recurrentes en las mujeres portadoras de esta anomalía (2,22). Es fundamental diferenciar un útero septado de un útero bicorne o didelfo, debido a las distintas opciones de tratamiento (3). La forma de diferenciarlos es mediante la visualización del contorno del fondo uterino, que tiene una convexidad externa lisa en el caso de un útero septado. La presencia de una hendidura mayor de 1 cm está relacionada con útero didelfo o bicorne (13,22). Asimismo es importante distinguir si el septo está compuesto por tejido fibroso, muscular (de similar señal a la del miometrio), o una combinación de ambas, ya que el primero puede ser reparado por histeroscopia, mientras que el segundo requiere un abordaje transabdominal (1,23). La longitud del septo no parece tener relevancia en los resultados obstétricos (22).
Clase VI. Útero arcuato. (Figura 10). Se manifiesta como una discreta indentación en el canal endometrial a nivel del fondo uterino, con un contorno externo normal, sin división de los cuernos uterinos. Esta anomalía es resultado de una reabsorción incompleta del septo útero-vaginal. Algunos autores la consideran una variante normal uterina, ya que no genera impacto en los resultados obstétricos de las pacientes portadoras de esta anomalía. Sin embargo, se puede considerar su reparación en casos de abortos a repetición (1,3,12). Clase VII. Anomalías asociadas al dietilestilbestrol (DES). El DES es un estrógeno sintético, usado hasta el año 1971. Este fármaco se asoció a adenocarcinoma de células claras de la vagina y múltiples alteraciones de las trompas, útero, cuello uterino y vagina, que alcanzaba hasta un 69% de las pacientes expuestas en etapas fetales al medicamento. Los clásicos hallazgos son hipoplasia uterina, cavidad endometrial en forma de T y bandas de constricción del fondo uterino, entre otras anormalidades encontradas (10,22).
CONCLUSIONES
La RM es una herramienta fundamental en la evaluación de la anatomía útero-vaginal y en el diagnóstico no invasivo de las anomalías müllerianas, permitiendo una adecuada caracterización y clasificación, para lo cual el radiólogo debe conocer la anatomía, embriología y clasificación de estas anomalías. El uso de imágenes multiplanares en relación al eje de un cuerpo uterino único o de los cuernos uterinos, es fundamental para la planificación quirúrgica. Asimismo permite diagnosticar los niveles de obstrucción en caso de tener endometrio funcionante, la presencia de un septo uterino caracterizando su longitud hacia caudal y la composición de éste.
BIBLIOGRAFÍA
1. Chandler T, Machan L, Cooperberg P, Harris A, Chang S. Müllerian duct anomalies: from diagnosis to intervention. Br J Radiol 2009;82:1034-42.
2. Propst A, Hill J. Anatomic factors associated with recurrent pregnancy loss. Semin Reprod Med 2000;18:341-50.
3. Junqueira B, Allen L, Spitzer R, Lucco K, Babyn P, Doria A. Müllerian duct anomalies and mimics in children and adolescents: Correlative intraoperative assessment with clinical imaging. Radiographics 2009;29:1085-103.
4. López-Clavijo C, de los Ríos L, Ibatá J. Prevalencia de anomalías müllerianas en un centro de medicina reproductiva. Rev CES Med 2012;26:155-64.
5. Chan Y, Jayaprakasan K, Zamora J, Thornton J, Raine- Fenning N, Coomarasamy A. The prevalence of congenital uterine anomalies in unselected and highrisk populations: a systematic review. Hum Reprod Update 2011;17:761-71.
6. Carrington B, Hricak H, Nuruddin R, Secaf E, Laros R, Hill E. Müllerian duct anomalies: MR imaging evaluation. Radiology 1990;176:715-20.
7. Pompili G, Munari A, Franceschelli G, Flor N, Meroni R, Frontino G, Fedele L, Cornalba G. Magnetic resonance imaging in the preoperative assesment of
Mayer-Rokitansky-Kuster-Hauser Syndrome. Radiol Med 2009;114(5):811-26.
8. Yoder I. Diagnosis of uterine anomalies: Relative accuracy of MR imaging, endovaginal sonography, and hysterosalpingography. Radiology 1992;185:343.
9. Saleem S. MR Imaging diagnosis of uterovaginal anomalies: current state of the art. Radiographics 2003; 23:e13-e13.
10. Díaz N, Riaño Y, Barón J, Ulloa L, Camargo C, Carrillo J. Anomalías de los conductos de Müller: una visión sencilla. Rev Colom Radiol 2008;19:2397-407.
11. Sadler TW. El aparato genitourinario. Langman Embriología Médica 11º ed. Barcelona: Editorial Lippincott Williams & Wilkins, 2009;235-63.
12. Troiano R, McCarthy S. Müllerian duct anomalies: imaging and clinical issues. Radiology 2004; 233:19-34.
13. Behr S, Courtier J, Qayyum A. Imaging of Müllerian Duct Anomalies. Radiographics 2012;32:E233–E250.
14. American Fertility Society-Birmingham Alabama. The American Fertility Society classification of adnexal adhesions, distal tubal occlusion, tubal occlusion secondary to tubal ligation, tubal pregnancies, Müllerian anomalies and intrauterine adhesions. Fertil Steril 1988;49:944-55.
15. Beguería R, Checa M, Castillo M, del Amo E, Carreras R. Malformaciones Müllerianas: clasificación, diagnóstico y manejo. Ginecol Obstet Clín 2009;10:165-9.
16. Yoo R, Cho J, Kim SY, Kim SH. Magnetic resonance evaluation of müllerian remnants in Mayer-Rokitansky- Küster-Hauser Syndrome. Korean J Radiol 2013;14:233-9.
17. Pomés C, Barrena N. Síndrome de Mayer-Rokitansky- Küster-Hauser: experiencia con vaginoplastía por tracción laparoscópica. Rev Chil Obstet Ginecol 2003;68:42-8.
18. Strübbe E, Willemsen W, Lemmens J, Thijn C, Rolland R. Mayer-Rokitansky-Küster-Hauser Syndrome: distinction between two forms based on excretory urographic, sonographic, and laparoscopic findings. AJR Am J Roentgenol 1993;160:331-4.
19. Oppelt P, Renner S, Kellermann A, Brucker S, Hauser G, Ludwig K, Strissel P, Strick R, Wallwiener D, Beckmann M. Clinical aspects of Mayer-Rokitansky Kuester- Hauser Syndrome: recommendations for clinicaldiagnosis and staging. Hum Reprod 2006;21:792-7.
20. Hall-Craggs M, Williams C, Pattison S, Kirkham A, Creighton S. Mayer-Rokitansky-Kuster-HauserSyndrome: diagnosis with MR imaging. Radiology 2013;269:787-92.
21. Khati N, Frazier A, Brindle K. The unicornuate uterus and its variants: clinical presentation, imaging and associated complications. J Ultrasound Med 2012;31:319-31.
22. Congenital Malformations of the Uterus. En: Hamm B., Forstner R. (eds). MRI and CT of the Female Pelvis. Medical Radiology. Springer Berlin Heidelberg, 2007;49-60.
23. Homer H, Li T, Cooke I. The septate uterus: a review of management and reproductive outcome. Fertil Steril 2000;73:1-14.