2018
1
Autores:
Francisco Fuentes Q1,, Eduardo Oliva A2, Carla Clavelle R3, Adriana Doren V, Sergio De la Fuente G
Imágen 1:
Imágen 2:
Imágen 3:
Tipo de documento:
Casos ClínicosDISPLASIAS ESQUELÉTICAS: REPORTE DE UN CASO Y REVISIÓN DE LA LITERATURA.
Contenido del documento:
RESUMEN
Las displasias esqueléticas son un grupo heterogéneo de condiciones que afectan primariamente la formación y crecimiento de huesos y cartílagos, se caracterizan por un acortamiento generalizado de huesos largos. Son patologías de baja prevalencia, que se pueden diagnosticar con precisión mediante ultrasonografía del primer y segundo trimestre. La importancia de esta patología radica en que posee una letalidad cercana al 50%. La displasia esqueletica letal más frecuente es la displasia tanatofórica, la cual se caracteriza por macrocefalia con base de cráneo estrecha, tórax estrecho, cuerpos vertebrales planos, micromelia generalizada, ausencia de fracturas, ventriculomegalia, polihidroamnios y mineralización ósea normal. Debido a que la presentación de la displasia tanatoforica se debe a una mutación autosómica dominante de novo no germinal, el riesgo de recurrencia no es mayor que el de la población general. Dado su elevada letalidad no pasa a generaciones futuras.
PALABRAS CLAVE: Displasia esquelética, ultrasonografìa, displasia tanatoforica
SUMMARY
Skeletal dysplasias are a heterogeneous group of conditions that primarily affect the formation and growth of bones and cartilage, characterized by a generalized shortening of long bones. These are pathologies of low prevalence, which can be accurately diagnosed by first and second trimester ultrasonography. The importance of this pathology lies in that it has a lethality close to 50%. The most common lethal skeletal dysplasia is tanophilic dysplasia, which is characterized by macrocephaly with a narrow cranial base, narrow chest, flat vertebral bodies, generalized micromelia, absence of fractures, ventriculomegaly, polyhydroamnios and normal bone mineralization. Because the presentation of the tanophoretic dysplasia is due to an autosomal dominant mutation of novo non-germinal, the risk of recurrence is not greater than that of the general population. Given its high lethality does not happen to future generations
KEY WORDS: Skeletal dysplasia, ultrasonography, tanophoretic dysplasia
Las displasias esqueléticas (DE) son un grupo heterogéneo de condiciones que afectan primariamente la formación y crecimiento de huesos y cartílagos (1-3). Junto a las disostosis, desórdenes óseos-metabólicos y síndromes reduccionales forman parte de una entidad denominada “desórdenes esqueléticos genéticos” (1).
Las DE son patologías de baja prevalencia, que se pueden diagnosticar con precisión mediante ultrasonografía (US) del primer y segundo trimestre (3-5). En conjunto comparten características básicas como el acortamiento generalizado de huesos largos, lo que se debe diferenciar de una restricción del crecimiento intrauterino (4, 5).
La importancia de esta patología radica en que si bien tiene una baja prevalencia, es responsable del 5% de los defectos congénitos encontrados en los recién nacidos (2) y posee una letalidad cercana al 50% (5, 6).
El avance de la US y los test moleculares con técnicas invasivas ha permitido no sólo un diagnóstico precoz, sino también una definición más precisa de letalidad. Ambos permiten también un mejor manejo y planificación de la vía de parto, disminuyendo tanto la morbilidad psicológica como física de la madre (1, 2, 7).
En este artículo se hará una revisión de las displasias esqueléticas y se presentará un caso clínico de una displasia letal (Tanatofórica).
Caso Clínico
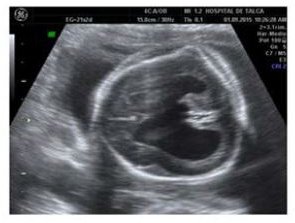
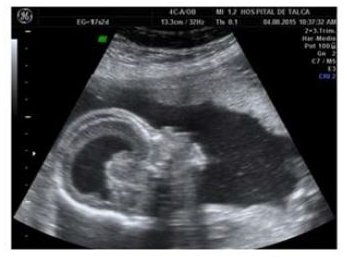
Paciente de 37 años, multípara de dos partos vaginales de término, con antecedente de Linfoma de Hodgkinen remisión completa hace 20 años, cursando un embarazo de 17 + 2 semanas por fecha de última regla (FUR) es derivada a comité ecográfico en el Hospital Regional de Talca por sospecha de síndrome malformativo. En la US se describe un feto único, vivo, con líquido amniótico normal, macrocráneo, ventriculomegalia y tórax estrecho (Figura 1 y 2). Debido a un severo acortamiento generalizado de huesos largos se plantea el diagnóstico de displasia esquelética.
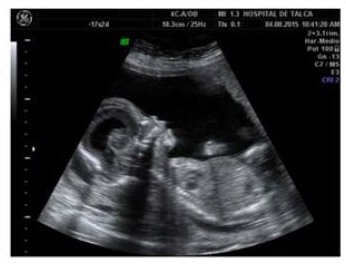
La paciente se mantiene en control en policlínico de alto riesgo obstétrico. En un nuevo examen ecográfico a las 21 semanas destaca: feto macrocráneo, con prominencia frontal y exoftalmos. También presentaba hiperextensión cervical, tórax estrecho para la edad gestacional y acortamiento severo y generalizado de huesos largos. El cráneo tenía forma de trébol (Figura 3 y 4) y había una marcada dilatación de los ventrículos cerebrales. Tras esta evaluación se diagnóstica “Displasia Esquelética Tanatofórica de tipo II”, y se mantiene en control en policlínico de alto riesgo.
Paciente inicia espontáneamente trabajo de parto a las 33+3 semanas. Por presentación podálica con feto con macrocefalia se realiza cesárea de urgencia, con histerotomía en T invertida debido a retención de cabeza última durante la cirugía. Se obtiene un recién nacido de sexo masculino, APGAR 1-1, que fallece a los 20 minutos de su nacimiento.
En el período postnatal inmediato se describe un recién nacido de 33 semanas, grande para la edad gestacional, cianótico, con frecuencia cardiaca de 60 lpm. Se observó macrocefalia con circunferencia craneana de 50 cm e hidrocefalia (Figura 5). Además se describen orejas de implantación baja, paladar ojival, cuello corto, tórax hipoplásico y plano, sin esfuerzo respiratorio, cordón umbilical con 3 vasos. Genitales masculinos con testes en escroto.
Discusión
Las displasias esqueléticas son un grupo heredable de más de 450 desórdenes bien definidos que afectan primariamente el tejido óseo y cartilaginoso, pero que también pueden afectar músculos, tendones y ligamentos (1-3). Fueron descritos por primera vez en 1960 como “Desórdenes constitucionales óseos” y se caracterizan por manifestarse en etapas tempranas de la gestación. Aunque estas anomalías son relativamente raras, poseen una prevalencia estimada de 2-5 por cada 10000 recién nacidos vivos, siendo responsable del 5% de los defectos congénitos (1, 2, 4).
Recientes avances en tecnología genética han permitido identificar las bases moleculares de más de 350 de estos desórdenes, dándonos la oportunidad de definir con precisión muchas de estas entidades, además de traducir los hallazgos de la investigación en servicios clínicos (1,2,5-8).
El diagnóstico prenatal se basa primariamente en la US del segundo trimestre, ya que el esqueleto fetal es fácilmente visualizado desde las 14 semanas (8-10). El diagnóstico debe ser confirmado idealmente por test moleculares a través de procedimientos invasivos, autopsia y radiografías post parto, incluyendo análisis histomórfico de cartílagos y huesos (1, 2). Cualquier feto que presente una longitud femoral o humeral bajo el percentil 5 o bajo 2 DS de la media para su EG debería ser evaluado de manera estandarizada, buscando otras anomalías en el esqueleto fetal (5, 6). La US permite un diagnóstico correcto en el 31-78% de los casos, y alcanza un 99% de precisión para definir fetos con displasias esqueléticas letales (5-10).
Un incremento en la translucencia nucal (TN) en la US del primer trimestre se ha asociado con displasia esquelética letal, lo que nos permite sospechar esta patología a una edad gestacional cada vez más temprana. Este incremento en la TN puede ser explicado por compresión mediastínica por la estrechez torácica y por las anormalidades en la composición de la matriz extracelular (defectos en el colágeno) (4-11).
La importancia de esta patología radica en que cerca del 50% de los casos son letales (23% mortinato, 32% mortalidad neonatal precoz). La prevalencia de displasia esquelética letal corresponde a 0.95 a 1.5 por 10000 RNV, siendo la displasia tanatofórica (DT) las más frecuente (29%) (1, 3, 4, 6).
Existen característica ecográficas que permiten definir la letalidad de una displasia esquelética. Éstas son: inicio temprano, micromelia severa, polihidroamnios, hidrops fetal no inmune y tórax estrecho. Éste último es el parámetro más importante para determinar letalidad, pues se traduce en hipoplasia pulmonar e insuficiencia respiratoria al nacer (2, 6).
Dentro de las DE letales la DT es la más frecuente, con una prevalencia de 0.24 a 0.69 por cada 10000 RNV. Fue descrita por primera vez en 1967 por Maroteaux y Lamy (11).Tanto hombres como mujeres son igualmente afectados. Junto a la acondroplasia, hipocondroplasia y la muy rara acondroplasia severa con desarrollo posterior de acantosis nigricans, pertenece al grupo de las osteocondrodisplasias de tipo 1 (2, 6, 10,13-15). Este grupo de desórdenes es producto de una mutación de novo en el receptor 3 del factor de crecimiento de fibroblastos (FGFR 3), el cual se localiza en el brazo corto del cromosoma 4 y es parte de la familia de los receptores de tirosina cinasa. La penetrancia de esta mutación es del 100% (13,16).Normalmente es un regulador negativo del crecimiento óseo y mutaciones puntuales aumentan su actividad enviando señales negativas a las células del cartílago (condrocitos), provocando una desorganización generalizada de la osificación encondral en la placa de crecimiento óseo. Debido a su letalidad esta mutación no pasa a las generaciones siguientes, salvo escasas excepciones (9,15,17,18).
La DT es casi uniformemente letal independiente del manejo. Los individuos afectados nacen muertos o mueren al cabo de algunas horas del parto por insuficiencia respiratoria, la que puede ser secundaria a hipoplasia pulmonar, compresión del tronco encefálico por el foramen magno estrecho o ambos (9,17).
Hallazgos ultrasonográficos característicos incluyen macrocefalia con base de cráneo estrecha, tórax estrecho, cuerpos vertebrales planos, micromelia generalizada, ausencia de fracturas, ventriculomegalia, polihidroamnios y mineralización ósea normal (6, 7, 9,13,14). Un estudio publicado en 2014 muestra que la identificación prenatal de displasia del lóbulo temporal podría ayudar en la identificación de DT, demostrado su presencia en el 67% de los casos analizados. Esto ha sido corroborado posteriormente por otros trabajos (17,19-21). La tomografía computada 3D prenatal contribuye al diagnóstico y podría tener un rol complementario a la US (22).
Se describen dos subtipos de DT, los cuales pueden ser diferenciados por la forma del fémur y el cráneo. La DT tipo I es la forma más frecuente (80%), usualmente causado por mutaciones R248C y Y373C del receptor 3 del factor de crecimiento de fibroblastos FGFR 3. Se caracteriza por un fémur en forma de “auricular de teléfono”, hipertelorismo, abombamiento frontal e hipoplasia del tercio medio facial, pero sin deformidad del cráneo (13,23-26). La DT tipo II se caracteriza usualmente por la mutación K650E en el gen FGFR3, sin embargo en 2015 se describió por primera vez una segunda mutación, llamada T394K (27). Sus características más específicas son fémures rectos y el cráneo en forma de hoja de trébol, esto dado por craneosinostosis prematura de las suturas lamboidea y coronal (9,10,23-25). De todas formas, se han notificado casos en que DT tipo I y II subtipos de superponen (13).
La DT puede afectar otros sistemas, observándose alteraciones como riñón en herradura, defecto septal atrial, defecto de válvula tricúspide, ano imperforado y sinostosis radiocubital (9,15).
Respecto a la consejería genética, debido a que la presentación de la DT se debe a una mutación autosómica dominante de novo no germinal, el riesgo de recurrencia no es mayor que el de la población general (13,25).
El diagnóstico prenatal de DT es sospechado por US, pero para su confirmación se sugiere solicitar análisis cromosómico (cariotipo) y de secuenciación de DNA, evaluando mutación FGFR3 en células fetales obtenidas por amniocentesis o biopsia de vellosidades coriales (9,13,17,26,28). Un estudio publicado en 2015 por Chitty et al sugiere que la secuenciación de próxima generación es más sensible que la reacción en cadena de polimerasa (PCR) (96.2 vs 88.6%) en diagnóstico prenatal para desordenes monogénicos, incluyendo DT y acondroplasia (29).
Dado que la acondroplasia es la forma más común de displasia esquelética no letal, se hace imprescindible su clara diferenciación con la DT. La acondroplasia, ocurre en 1 in 15,000 a 40,000 RNV (2,9,30). Al igual que la DT es causada por mutación en el gen FGFR3. La mayoría de los casos se diagnostican al nacer, aunque en algunos niños se sospecha la patología recién en la primera infancia. Se caracteriza por acortamiento de predominio rizomélico de aparición tardía (>24 semanas), macrocefalia, abombamiento frontal, depresión puente nasal y manos en tridente. Presenta un patrón de herencia autosómico dominante. Los individuos que heredan dos copias alteradas de este gen típicamente tienen una forma severa de acondroplasia que causa acortamiento extremo de los huesos y una caja torácica subdesarrollada. Estos individuos suelen fallecer poco después del nacimiento debido a insuficiencia respiratoria. La acondroplasia heterocigota es compatible con una vida, siendo la DE más frecuente en pacientes que llegan a la adultez (9,30).
BIBLIOGRAFÍA
1. Warman M, Cormier-Daire, Hall C, Kravow D, Lachman R, LeMerrer M, et al. Nosology and Classification of Genetic Skeletal Disorders: 2010 Revision. Am J Med Gen 2010;155:943-968.
2. Krakow D. Skeletal Dysplasias. Clin Perinatol 2015;42: 301–319.
3. Goncalves L, Espinoza J, Mazor M, Romero R. Newer imaging modalities in the prenatal diagnosis of skeletal dysplasias. Ultrasound Obstet Gynecol 2004; 24: 115–120.
4. Ngo C, Viot G, Aubry M, Tsatsaris V, Grange G, Cabrol D, Pannier E. First-trimester ultrasound diagnosis of skeletal displasia associated with increased nuchal translucency thickness. Ultrasound Obstet Gynecol 2007; 30: 221–226.
5. Krakow D, Lachman R, Rimoin D. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet Med 2009;11:127-133.
6. Schramm T, Gloning K, Minderer S, Daumer-Haas C, Hortnagel K, Nerlich A, et al. Prenatal sonographic diagnosis of skeletal dysplasias. Ultrasound Obstet Gynecol 2009; 34: 160–170
7. Jeanty P, Valero G. La valoración del feto con displasia esquelética. Women’s Health Alliance, Nashville, TN, USA and * Ultrasonido Diagnóstico, Magdalena, Son, México.
8. Lachman, RS. Taybi and Lachman’s radiology of syndromes, metabolic disorders and skeletal dysplasias. 5th ed. St. Louis, MO: Mosby; 2007.
9. Giraldo-Cuartas A. Displasia Tanatofórica, reporte de un caso y revisión. Revista Colombiana de Obstetricia y Ginecología. 2008; 59(4); 349-356.
10. Phyllis Glanc, et al. Prenatal diagnosis of the lethal skeletal dysplasias. UpToDate. Jun 2015.
11. Khalil A, Pajkrt E, Chitty L. Early prenatal diagnosis of skeletal anomalies. Prenat Diagn. 2011; 31: 115–124.
12. Maroteaux P, Lamy M, Robert JM. Thanatophoric dwarfism. Presse Med 1967;75:2519-2524.
13. Defendi G. Thanatophoric Dysplasia. Pediatrics: Genetics and Metabolic Disease 2016. Visitado en 2017 Jan 13. Disponible en: http://emedicine.medscape.com/article/949591-overview.
14. Padilla A, Durán M, Davies B. Displasia tanatofórica: revisión de los criterios de diagnóstico en 5 casos de autopsia. Rev Mex Pediatr 2005; 72 (3):126-132.
15. Nikkel S, Major N, King W. Growth and development in thanatophoric displasia – an update 25 years later. Clinical Case Reports 2013; 1(2): 75–78.
16. Martinez M, de Frutos C, Nieto A. Review of the Recently Defined Molecular Mechanisms Underlying Thanatophoric Dysplasia and Their Potential Therapeutic Implications for Achondroplasia. Am J Med Genet 2009;15:245–255.
17. Naveen N, Murlimanju B, Kumar V, Palakunta T, Jeeyar H. Thanatophoric Dysplasia: A Rare Entity. Oman Medical Journal 2011;26(3): 196-197.
18. Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocr Rev 2000; 21(1): 23–39.
19. Wang D, Shannon P, Toi A, Chitayat D, Mohan U, Barkova E, et al. Temporal lobe dysplasia: a characteristic sonographic finding in thanatophoric displasia. Ultrasound Obstet Gynecol. 2014;44(5):588-594.
20. Pazzaglia U, Donzelli C, Izzi C, Baldi M, Di Gaetano G, Bondioni M. Thanatophoric dysplasia. Correlation among bone X-ray morphometry, histopathology, and gene analysis. Skeletal Radiol 2014;43:1205–1215.
21. Wang D, Shannon P, Toi A, Chitayat D, Mohan U, Barkova E, et al. Temporal lobe dysplasia: a characteristic sonographic finding in thanatophoric displasia. Ultrasound Obstet Gynecol 2014; 44: 588–594.
22. Ulla M, Aiello H, Cobos M, Orioli I. Prenatal Diagnosis of Skeletal Dysplasias: Contribution of Three-Dimensional Computed Tomography. Fetal Diagn Ther 2011;29:238–247.
23. Chen CP, Chern SR, Shih JC, Wang W, Yeh LF, Chang TY, Tzen CY. Prenatal diagnosis and genetic analysis of type I and type II thanatophoric dysplasia. Prenat Diagn 2001; 21(2): 89-95.
24. Langer LO Jr, Yang SS, Hall JG, Sommer A, Kottamasu SR, Golabi M, Krassikoff N. Thanatophoric dysplasia and cloverleaf skull. Am J Med Genet Suppl 1987; 3:167-179.
25. Genetics Home Reference, published January 10 2017. Visitado en 2017 Jan 13. Disponible en: https://ghr.nlm.nih.gov/condition/thanatophoric-dysplasia.
26. Sharma M, Jyoty, Jain R, Devendra. Thanatophoric Dysplasia: A Case Report. Journal of Clinical and Diagnostic Research 2015;9(11):01-03.
27. Gulasi S, Atici A, Celik Y. A Case of Thanatophoric Dysplasia Type 2: A Novel Mutation. J Clin Res Pediatr Endocrinol 2015;7(1):73-76.
28. Jung E, Wook H, Nam K, Yeon S. A case of thanatophoric dysplasia type I with an R248C mutation in the FGFR3 gene. Korean J Pediatr 2010;53(12):1022-1025.
29. Chitty L, Mason S, Barrett A. Non-invasive prenatal diagnosis of achondroplasia and thanatophoric displasia: next-generation sequencing allows for a safer, more ccurate, and comprehensive aprroach. Prenat Diagn 2015;35(7):656-662.
30.Genetics Home Reference, published January 10 2017. Visitado en 2017 Jan 14. Disponible en: https://ghr.nlm.nih.gov/condition/achondroplasia.


