2010
4
Autores:
Instituciones:
Ana Isabel Padilla P., Margarita Álvarez de la Rosa R., Marina Rodríguez M., Yanet Carballo L., Francisco de la Torre F., Juan Troyano L
Imágen 1:
Imágen 2:
Imágen 3:
Tipo de documento:
Casos ClínicosHIDROPS FETAL COMO MANIFESTACIÓN ECOGRÁFICA DE UNA ENFERMEDAD FAMILIAR: INCONTINENTIA PIGMENTI (SÍNDROME DE BLOCH-SULZBERGER)
Contenido del documento:
RESUMEN
Diagnóstico de un caso de enfermedad familiar a partir de un hidrops fetal. Gestante con 2 abortos previos ocurridos al inicio del segundo trimestre, ambos fetos masculinos. Se detecta en la semana 12 un higroma quístico en el contexto de un hidrops fetal. Estudio serológico (TORCH y parvovirus B12), con resultado negativo y biopsia corial con cariotipo 46 XY. La anamnesis y exploración detallada hacen sospechar una enfermedad ligada al cromosoma X, dado que la paciente y una hermana están afectas de una genodermatosis desde la infancia: Incontinentia Pigmenti.
PALABRAS CLAVE: Incontinentia pigmenti, abortos de repetición, hidrops fetal
SUMMARY
We revealed a family disease by a fetal anomaly. The patient had a history of two early miscarriages of male fetuses. In the present pregnancy hydrops fetalis was demonstrated by routine ultrasound scan at 12 weeks. Serological tests were negative and chorionic villous sampling revealed normal karyotype, 46XY. We suspected a X-linked disease. Detailed family history leaded to the demonstration that the patient and a sister were affected of a rare genodermatosis: Incontinentia Pigmenti.
KEY WORDS: Incontinentia Pigmenti, habitual abortion, hydrops fetalis
INTRODUCCIÓN
La Incontinentia Pigmenti (IP, OMIM 308300) es una genodermatosis que puede estar asociada con una gran variabilidad de alteraciones en ojos, uñas, pelo, dientes, esqueleto, corazón y retraso mental. Se transmite de forma autosómica dominante ligada al cromosoma X, por lo que es letal en embriones varones. Las mujeres afectas sobreviven gracias a la duplicidad del cromosoma X y a la inactivación temprana del que aporta la mutación, mientas que los varones que han heredado el gen mutado normalmente fallecen. La mutación que da lugar a la IP es una delección de gran parte del gen NEMO (1). Las alteraciones cutáneas pueden estar presentes en el momento del nacimiento o aparecer en los primeros 15 días de vida, con el tiempo, se van atenuando, llegando a desaparecen en la pubertad y fase adulta. La dificultad del diagnóstico, como en el caso que se presenta, radica en que el cuarto estadio de la enfermedad es una manifestación tardía, probablemente infradiagnosticada, que consiste en alteraciones tróficas hipopigmentadas en los miembros inferiores. En algunos casos quedan lesiones neurológicas residuales como retraso mental y/o graves afectaciones motoras y oculares.
El hidrops embrionario ocurre en múltiples situaciones siendo las más características las aneuploidías y las infecciones. El hallazgo de un hidrops precoz con cariotipo normal y serologías negativas puede ser indicativo de una enfermedad autosómica dominante o ligada al X (2).
En muy pocas ocasiones se ha descrito el diagnóstico de la IP familiar a partir de un embrión afectado (3). En el presente caso el cuadro apareció como abortos recurrentes de fetos varones en una paciente con una forma subclínica no diagnosticada de la enfermedad y una hermana con retraso mental. Este diagnóstico permite ofrecer consejo genético a las parejas, e incluso conseguir la gestación de un embrión sano mediante técnicas de reproducción asistida (4).
Caso clínico
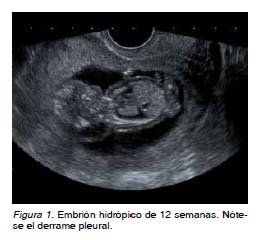
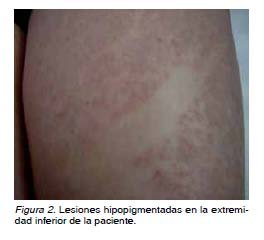
Se trata de una gestante de 38 años de edad, sin historial de consanguinidad, Rh positivo y con esterilidad primaria. Es remitida con un embarazo conseguido mediante técnicas de reproducción asistida a la Unidad de Ecografía en la semana 12, por translucencia nucal patológica. La paciente refiere dos abortos previos de 13 semanas y ambos de sexo masculino. En la ecografía de control en la semana 12 aparece una gestación única, intrauterina, activa, acorde a 12 semanas. La translucencia nucal es de 5 mm, de aspecto tabicado, en el contexto de un hidrops masivo (hidrotórax y ascitis), sin objetivarse otras alteraciones estructurales (Figura 1). Se solicita entonces estudio serológico, que fue negativo para toxoplasma, rubéola, citomegalovirus, herpes virus y parvovirus B19, y se aconseja cariotipo fetal mediante biopsia corial, donde se confirma que se trata de un embrión de sexo masculino (46XY). Mediante anamnesis exhaustiva, la paciente refiere y se objetivan lesiones hipopigmentadas en sus miembros inferiores acompañadas de ausencia de vello (Figura 2).
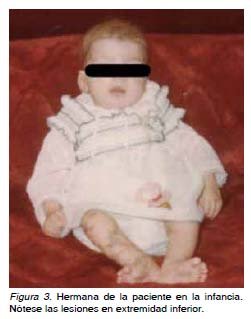
Tras la valoración de los antecedentes familiares de posibles enfermedades o patologías que tuviesen carácter hereditario, se revela la sospecha de un trastorno que pudiese estar ligado al cromosoma X. La madre de la paciente tuvo 2 abortos tardíos (fetos varones), además la paciente tiene una única hermana con retraso mental, que al nacimiento presentó una dermatosis que, como en su caso, dejó las secuelas claras de hipopigmentación en miembros inferiores, se le solicita documentación de la misma que la paciente aporta (Figura 3). Se procede entonces, a cursar una interconsulta con el Servicio de Dermatología quienes confirman el diagnóstico de IP. La gestación se interrumpe de forma espontánea, se realiza legrado, y a la pareja se le indica la posibilidad de conseguir una próxima gestación mediante técnicas de reproducción asistida, como el diagnóstico preimplantacional con selección de sexo, o bien, la elección de ovocitos de donante. En la actualidad la paciente se encuentra gestante de un embarazo gemelar bicorial obtenido mediante donación de ovocitos.
DISCUSIÓN
Se ha diagnosticado una enfermedad familiar a partir de un hidrops embrionario y el estudio de la familia. La IP es una genodermatosis ligada al X asociada con una gran variabilidad de alteraciones en ojos, uñas, pelo, dientes, esqueleto, corazón y sistema nervioso central. Se han descrito pocos casos de hidrops fetal ecográfico (5). Las mujeres afectas sobreviven gracias a la inactivación temprana del cromosoma X que aporta la mutación. Los varones que han heredado el gen mutado normalmente no sobreviven.
La mayoría de los hidrops son de tipo no inmune, representando el 76-87% y las causas principales del mismo son: patología cardiovascular (21,7%), cromosómicos (13,1%), hematológicos (10,4%), sindrómicos (4,4%), infecciones (6,7%), malformaciones torácicas (5%), malformaciones urinarias (2,3%), síndrome de transfusión feto-fetal (5,6%) e idiopáticos (17,8%) (2). Entre estos últimos se encontrarían las enfermedades genéticas como probable etiología.
En el caso descrito se llegó al diagnóstico de la causa de hidrops tras descartar otras causas y mediante investigación familiar. La incidencia de IP es de 1:40.000 nacimientos, pudiendo asociar procesos neoplásicos como la leucemia mielógena, el tumor de Wilms o el rabdomiosarcoma. Por tanto, se trata de una enfermedad potencialmente grave que obliga a un seguimiento multidisciplinar del paciente afectado e impone el consejo genético.
Por tanto, cuando se descartan el origen anatómico, endocrino, infeccioso o inmunológico del hidrops, se debe canalizar a la pareja al genetista. El especialista, a través de una detallada historia clínica, podrá determinar si existen factores de riesgo.
Existen pocos artículos en la literatura que asocien hidrops fetal con el diagnóstico precoz o tardío de esta enfermedad (5-8). Ya se ha descrito la selección embrionaria mediante técnicas de reproducción asistida (4) y en la forma familiar de la enfermedad está disponible el diagnóstico molecular mediante biopsia de vellosidades coriales, ya que se ha localizado la enfermedad en la región Xq28 (9).
CONCLUSIÓN
En casos de abortos recurrentes se debe investigar cuidadosamente cualquier indicio de enfermedad familiar.
BIBLIOGRAFÍA
BIBLIOGRAFÍA
1. Berlin AL, Paller AS, Chan LS. Incontinentia pigmenti: a review and update on the molecular basis of pathophysiology. J Am Acad Dermatology 2002;47:169-87.
2. Bellini C, Hennekam RC, Fulcheri E, Rutigliani M, Morcaldi G, Boccardo F, Bonioli E. Etiology of noimmune hydrops fetalis: a systematic review. Am J Med Genet A 2009;149A:844-55.
3. Robyr R, Bernard JP, Roume J, Ville Y. Familial diseases revealed by a fetal anomaly. Prenat Diagn 2006;26:1224-34.
4. Pettigrew R, Kuo HC, Scriven P, Rowell P, Pal K, Handyside A, et al. A pregnancy following PGD for X-linked dominant [correction of X-linked autosomal dominant] incontinentia pigmenti (Bloch-Sulzberger syndrome): case report. Hum Reprod 2000;15:2650-2.
5. Dufke A, Vollmer B, Kendziorra H, Mackensen-Haen S, Orth U, Orlikowsky T, et al. Hydrops fetalis in three male fetuses of a female with incontinentia pigmenti. Prenat Diagn 2001;21:1019-21.
6. Turnpenny PD, Gunasegaran R, Smith NC, Dean JC. Recurrent miscarriage, cystic hygroma and incontinentia pigmenti. Br J Obstet Gynaecol 1992;99:920-1.
7. Odent S, Le Marec B, Smahi A, Hors-Cayla C, Milon J, Jouan H, Laurent MC, Borg AM. [Spontaneous abortion of male fetuses with incontinentia pigmenti (apropos of a family)]. J Gynecol Obstet Biol Reprod (Paris) 1997;26:633-6.
8. Devriendt K, Matthijs G, Fryns JP, Ballegeer V. Second trimester miscarriage of a male fetus with incontinentia pigmenti. Am J Med Genet 1998;80:298-9.
9. Le Roux BP, Hornez G, Beaulieu P, Darie H, Millet P. [Incontinentia pigmenti]. Arch Pediatr 1996;3:152-5.


