2020
4
Autores:
Laura García-Calvo 1, Covadonga Martí Álvarez 1,2, Polán Ordás Álvarez 1, Elena Sara Merino 1, Laura Frías Aldaguer 1,2, Óscar Cerezo Aranda 3, J.Ignacio Sánchez Méndez 1,2.
Instituciones:
1 Servicio de Ginecología y Obstetricia. Hospital Universitario La Paz, Madrid España.
2 Unidad de Patología Mamaria. Hospital Universitario La Paz, Madrid España.
3Servicio de Anatomía Patológica. Hospital Universitario La Paz, Madrid, España.
Correspondencia: Laura García Calvo, laugarca.detec@gmail.com.
Tipo de documento:
Casos ClínicosPlasmocitoma mamario y cáncer de mama. A propósito de un caso.
Contenido del documento:
INTRODUCCIÓN
El plasmocitoma se engloba dentro de un grupo de entidades caracterizadas por una proliferación descontrolada de un clon celular de células plasmáticas. Estas neoplasias se pueden presentar como una lesión aislada (plasmocitoma solitario), o en forma de mieloma múltiple (MM). Los plasmocitomas solitarios típicamente aparecen en el hueso pero también pueden desarrollarse en otros tejidos (plasmocitoma extramedular). Aún se desconocen las razones que implican el desarrollo en algunos pacientes de la forma aislada de la enfermedad y en otros de la forma diseminada (MM).
Una de estas presentaciones extramedulares es el plasmocitoma mamario. Se trata de una entidad extremadamente rara, con menos de cincuenta casos descritos en la literatura en el último siglo (1). Por este motivo, disponemos de pocos datos acerca de su abordaje y tratamiento óptimos. Además, el seguimiento es variable y no conocemos su evolución a largo plazo.
El interés del caso expuesto radica tanto en su abordaje terapéutico mediante cirugía como en el desarrollo ulterior de un cáncer de mama, cuyo tratamiento podría haber estado condicionado por el manejo previo del plasmocitoma
CASO CLÍNICO
Presentamos el caso de una paciente de 70 años, sin antecedentes médicos de interés excepto glaucoma y trombosis de la arteria retiniana derecha, e intervenida de faquectomía bilateral. Acudió al Servicio de Dermatología por una lesión única eritematosa nodular en la mama derecha (FIGURA 1), de casi dos años de evolución, no dolorosa, de aspecto eritematodescamativo de 2 x 1.5 cm. Se realizó un punch de la lesión, que fue informado como plasmocitoma/mieloma (a dependencia del contexto clínico).
En el estudio anatomopatológico se observó una proliferación “en sábana” de células plasmocitoides, que ocupaban prácticamente toda la dermis y parte de la hipodermis, respetando la epidermis (FIGURA 2).
Ante una proliferación de hábito plasmocitoide en piel de mama, se debe descartar como primera opción un carcinoma de mama o un melanoma. Esto se realiza con tinciones específicas (CD 138, MelanA, cadenas ligeras kappa/lambda etc) (FIGURAS 3-8). Si se excluyen estas entidades la siguiente posibilidad sería un linfoma, y dada la morfología celular de nuestra paciente, un plasmocitoma.
La paciente fue remitida al Servicio de Hematología, descartándose, tras un estudio completo con biopsia de Médula ósea, PET-TC, analítica y proteinograma en orina y en sangre, que se tratara de una lesión en el contexto de un mieloma múltiple. Una vez confirmada la existencia de una lesión única se presentó el caso en el Comité Multidisciplinar de Patología Mamaria donde se decidió tratamiento quirúrgico.
Se llevó a cabo una tumorectomía con márgenes sin incidencias. Tras seis meses de seguimiento, se realiza mamografía de revisión en la que se visualiza una tenue distorsión con microcalcificaciones en CSE de la mama derecha que había aumentado respecto a estudios previos (FIGURA 9). Ante la sospecha de malignidad se lleva a cabo una ecografía mamaria que encuentra un área hipoecoica mal delimitada con flujo vascular de tres centímetros de diámetro máximo, con axila ecográficamente negativa. Se efectúa una biopsia con aguja gruesa (BAG) en dicha zona, con un diagnóstico anatomopatológico de carcinoma lobulillar infiltrante G2 con receptores estrogénicos positivos 100 %, receptores de progesterona positivos 60 %, Ki67 10 %, citoqueratina 19 positiva, Cadherina E negativa y sin sobreexpresión del oncogén HER2/neu.
La paciente recibió hormonoterapia neoadyuvante 4 meses consiguiendo una respuesta fragmentada parcial. Posteriormente se realizó una tumorectomía guiada por arpón con biopsia selectiva de ganglio centinela, que fue positivo, por lo que se completó con linfadenectomía axilar derecha. Tras realizarse plataforma genómica (Endopredict/EP-clin) con resultado de bajo riesgo, se completó tratamiento adyuvante con radioterapia en mama y axila (48 Gy) y permanece libre de enfermedad tras 6 meses de seguimiento.
DISCUSIÓN
El plasmocitoma cutáneo es una entidad muy poco frecuente. La forma de presentación más habitual es en el contexto de un mieloma múltiple, el cual solo en un 2-4% de los casos presenta afectación cutánea. La forma primaria es extremadamente rara y es preciso realizar un estudio de extensión, para poder descartar la existencia de un mieloma subyacente.
La forma típica de presentación del plasmocitoma cutáneo es similar a la de nuestra paciente: nódulo subcutáneo, asintomático de años de evolución, de predominio en tronco y que histológicamente corresponde a una proliferación de células plasmáticas con poca atipia. Suele presentarse en pacientes adultos, con una edad media de 53 años (2, 3) y se asocia en torno a un 60-70% con la presencia de mieloma múltiple.
El diagnóstico anatomopatológico se realiza mediante la visualización de una proliferación de células plasmáticas junto con los marcadores inmunohistoquímicos específicos, como se realizó en nuestro caso.
Otros autores han utilizado otras técnicas. La técnica de hibridación fluorescente in situ (FISH) se ha realizado en dos pequeños estudios con plasmocitomas extramedulares donde se ha identificado una alta incidencia de pérdida 13q (de un 33 a un 40%), translocaciones en las Inmunoglobulinas de cadena pesada (en aproximadamente 37 al 53%) e hiperploidía en el 54% de sus casos (4,5).
Dos son fundamentalmente las alternativas terapéuticas de los plasmocitomas extramedulares: la radioterapia y la cirugía, pudiendo plantearse otras alternativas en casos recurrentes.
Es bien conocido que los plasmocitomas son radiosensibles, con una tasa de respuesta del 79-90% y una tasa de supervivencia del 50-100% (1,6-8). La enfermedad extramedular aislada tiene mejor pronóstico que el plasmocitoma óseo y se puede curar solo con radioterapia local (1,6).Por otro lado, dado que la mayoría de los plasmocitomas extramedulares (EMP) se dan en la zona de cabeza y cuello y la cirugía radical con intención curativa conlleva procedimientos muy agresivos o mutilantes, la radioterapia se convierte en la opción ideal (1-6,8-10). Uno de los pocos casos publicados tratados con RT es el de Majadob K. (7), en su paciente de 29 años con un plasmocitoma mamario en octubre de 2009, se le trató con radioterapia radical (40.05 Gy fraccionado en tres semanas).
En pacientes con EMP de localización accesible quirúrgicamente, la cirugía con fines curativos es otra de las alternativas considerada por algunos autores, quedando la radioterapia relegada a los casos de afectación de márgenes (11).
Algunos autores como Kilciksiz han abogado por la combinación de cirugía con dosis moderadas de radioterapia (12-14), reportando mejor pronóstico en estos casos que en los tratados únicamente con radioterapia, aunque con tiempos de seguimiento cortos. Esto contradice otros estudios que defienden la radioterapia aislada como tratamiento (15), o el ya mencionado trabajo de Majadob K. Luca y colaboradores describen también la realización de ambos tratamientos con resultados satisfactorios. La mayoría de los pacientes de su serie permanecieron sin enfermedad ni manifestaciones sistémicas de mieloma durante años (17).
En el caso que presentamos, tras valoración en comité multidisciplinar, se optó por la cirugía, dada la accesibilidad de la lesión y su pequeño tamaño. Puesto que se consiguieron márgenes libres, la radioterapia quedó definitivamente desestimada.
La posibilidad de recurrencia del plasmocitoma es uno de los puntos cruciales de esta enfermedad. La tasa de recaída varía según distintos autores, aunque la mayoría de los trabajos publicados corresponden a series pequeñas de pacientes. Tras tratamiento con radioterapia con o sin quimioterapia, el porcentaje de recurrencias locales puede ascender a un 25% de los pacientes (18). Las metástasis extramedulares a distancia no son excepcionales, en un 30 a un 50% de los casos la enfermedad progresa hacia un mieloma múltiple en un tiempo medio de 1.5 a 2.5 años (19). Sin embargo, en otras series, la tasa de recurrencia local tras radioterapia fue menor al 5% (12), siendo el riesgo de enfermedad a distancia del 30%.
El tratamiento de las recaídas también es relativamente heterogéneo. De acuerdo a las guías British Committee for Standars in Haematology (BCSH) de 2009, la recurrencia del plasmocitoma aislado en el sitio original de la radioterapia, en ausencia de enfermedad sistémica, puede ser tratada con radioterapia adicional. Los pacientes con enfermedad sistémica o recaída temprana, pueden beneficiarse de terapia sistémica añadiendo o no transplante autólogo de médula ósea, al igual que está indicado en el mieloma. Otros agentes como talidomida y bortezomid se han demostrado útiles antes de transplante, en un pequeño número de pacientes con recaída de plasmocitoma.
Nuestra paciente no ha presentado recaída del plasmocitoma, aunque el seguimiento ha sido tan sólo de algo más de un año. Sin embargo, fue diagnosticada de un carcinoma lobulillar ipsilateral. No hay otros casos de carcinoma de mama tras plasmocitoma en la literatura, y, por tanto, no hay datos que permitan establecer una relación entre ambos. Sin embargo, el hecho de haber manejado quirúrgicamente el plasmocitoma, prescindiendo de la radioterapia, permitió realizar una cirugía conservadora que no hubiera sido posible si dicha mama hubiera estado radiada.
CONCLUSIÓN E IMPORTANCIA CLÍNICA
Se trata del primer caso descrito en la literatura del desarrollo de un carcinoma de mama en una paciente con un plasmocitoma mamario previo, lo que nos reafirma en la necesidad de un seguimiento estrecho sin olvidar la posibilidad de aparición de una neoplasia de novo al igual que en cualquier mujer. Además, en estos casos, es importante plantear el tratamiento quirúrgico como primera opción pues eso no alteraría las opciones terapéuticas en caso de aparición ulterior de un cáncer de mama.
REFERENCIAS
- Kaviani A, Djamali-Zavareie M, Noparast M, Keyhani-Rofagha S. Recurrence of primary extramedullary plasmacytoma in breast both simulating primary breast carcinoma. World J Surg Oncol. 2004;2(1):29.
- Ross JS, King TM, Spector JI, Zimbler H, Basile RM. Plasmacytoma of the breast. An unusual case of recurrent myeloma. Arch Intern Med. 1987;147(10):1838–1840.
- Taylor L., Aziz M., Klein P., Jagannath S., Axelrod D. Plasmacytoma in the breast with axillary lymph node involvement: a case report. Clinl Breast Cancer. 2006;7(1):81–84.
- Bink K, Haralambieva E, Kremer M, Ott G, Beham-Schmid C, de Leval L, Peh SC, Laeng HR, Jütting U, Hutzler P, et al. Primary extramedullary plasmacytoma: similarities with and differences from multiple myeloma revealed by interphase cytogenetics. Haematologica. 2008;93:623–626. doi: 10.3324/haematol.12005.
- Boll M, Parkins E, O’Connor SJM, Rawstron AC, Owen RG. Extramedullary plasmacytoma are characterized by a ‘myeloma-like’ immunophenotype and genotype and occult bone marrow involvement. Br J Haematol. 2010;151:525–527. doi: 10.1111/j.1365-2141.2010.08386.x.
- Wiltshaw WE. The natural histology of extramedullary plasmacytoma and its relation to solitary myeloma of bone and myelomatosis. Medicine Baltimore. 1979;55:217–238.
- Majadob K, Al-Sakkaf W, Rezk F, Zegocki K, Al-Refaie F. Recurrence of extramedullary plasmacytoma of the breast. Ecancermedicalscience. 2013;7:322. Published 2013 May 28. doi:10.3332/ecancer.2013.322
- Surov A, Holzhausen HJ, Ruschke K. Breast plasmacytoma. 2010;51(5):498–504. doi: 10.3109/02841851003712924.
- De Chiara A, Losito S, Terracciano L, Di Giacomo R, Laccarino G, Rubolotta M. Primary plasmacytoma of the breast. Arch Pathol Lab Med. 2001;125:1078–80.
- Dimopoulos MA, Kiamouris C, Moulopoulos LA. Solitary plasmacytoma of bone and extramedullary plasmacytoma. Hematology. 1999;13(6):1249–1257.
- Alexiou C, Kau RJ, Dietzfelbinger H, et al. Extramedullary plasmacytoma: tumor occurrence and therapeutic concepts. Cancer. 1999;85(11):2305.
- Kilciksiz S, Karakoyun-Celik O, Agaoglu FY, Haydaroglu A. A review for solitary plasmacytoma of bone and extramedullary plasmacytoma. ScientificWorldJournal. 2012;2012:895765. doi:10.1100/2012/895765
- Liebross RH, Ha CS, Cox JD, Weber D, Delasalle K, Alexanian R. Clinical course of solitary extramedullary plasmacytoma. Radiother Oncology. 1999;52:245–9. doi: 10.1016/S0167-8140(99)00114-0.
- Tsang RW, Gospodarowicz MK, Pintilie M, et al. Solitary plasmacytoma treated with radiotherapy: impact of tumor size on outcome. International Journal of Radiation Oncology Biology Physics. 2001;50:113–120.
- Jyothirmayi R, Gangadharan VP, Nair MK, Rajan B. Radiotherapy in the treatment of solitary plasmacytoma. British Journal of Radiology. 1997;70:511–
- Kilciksiz S, Celik OK, Pak Y, et al. Clinical and prognostic features of plasmacytomas: a multicenter study of Turkish Oncology Group—Sarcoma Working Party. American Journal of Hematology. 2008;83(9):702–707.
- Luca G, Rawn S, Indira G, Lisa J, Robert S. Plasmacytoma of the breast. Journal of Family practice. 2011;20:86–88.
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed. Lyon, France: IARC, 2008; Vardiman JW (Eds.)
- Park YM. Imaging findings of plasmacytoma of both breasts as a preceding manifestation of multiple myeloma. Case Rep Med. 2016:6595610.
FIGURAS
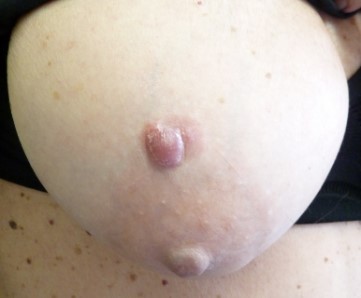
Figura 1. Lesión mama derecha.
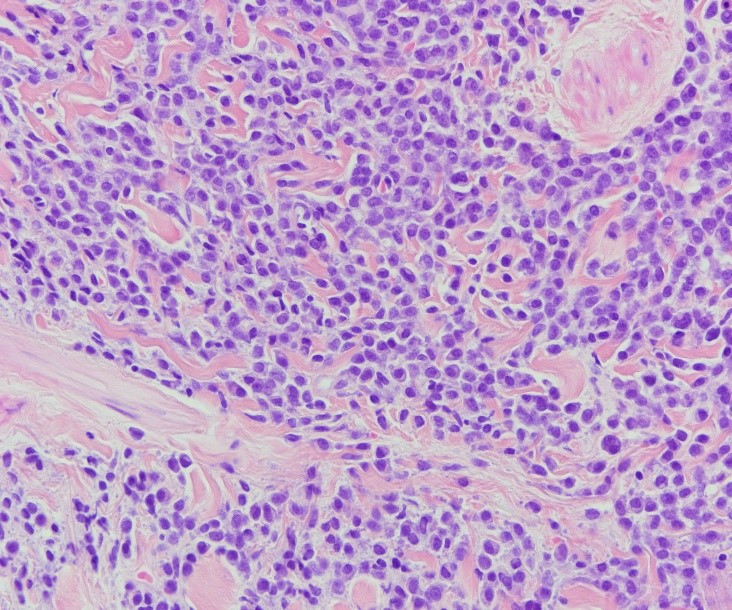
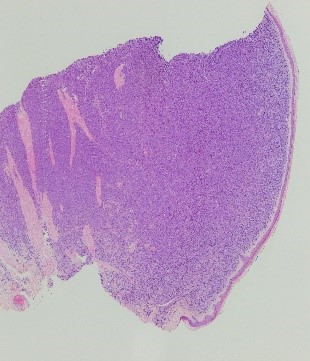
Figura 2. Hábito plasmocitoide.
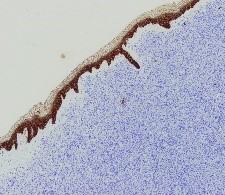
Figura 3. Tinción CK: citoqueratina. Marcador de célula de estirpe epitelial, negativa.
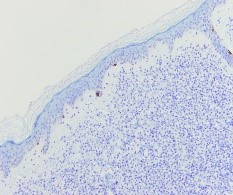
Figura 4. Tinción Melan A negativa, de diferenciación melanocítica.
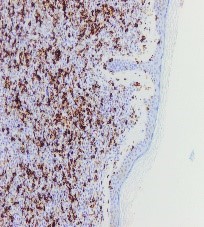
Figura 5. CD 45. Marcador linfocitario negativo.
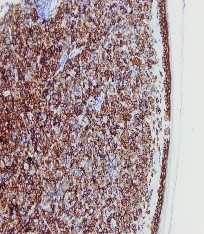
Figura 6. CD 138 positivo, en: cc plasmáticas, neg en cc hematopoyéticas y endoteliales.
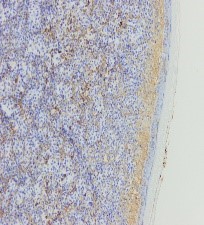
Figura 7. Kappa negativo
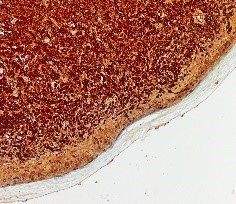
Figura 8. Lambda monoclonal
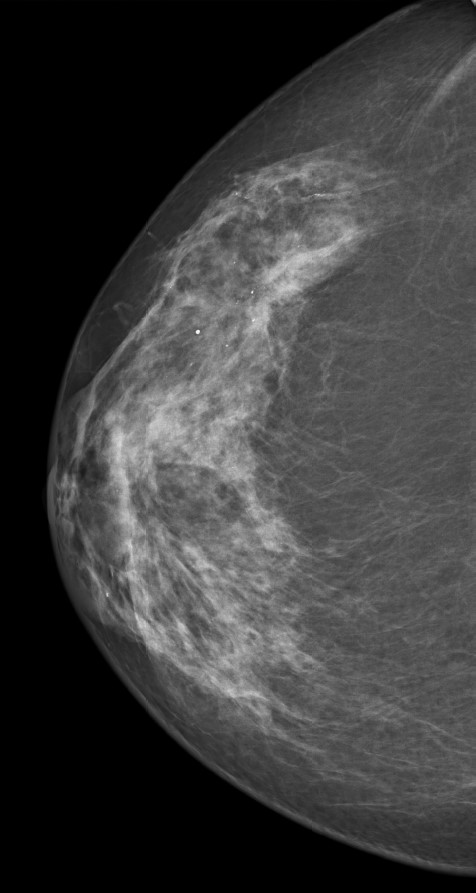
Figura 9. Microcalcificaciones en CSE MD.


