2011
1
Autores:
Instituciones:
Wilmar Saldarriaga Gil, Fernando Ávila Sánchez, Carolina Isaza de Lourido.
Imágen 1:
Imágen 2:
Imágen 3:
Tipo de documento:
Casos ClínicosSÍNDROME DE TURNER CON MOSAICISMO 45X/46XY: REPORTE DE CASO
Contenido del documento:
RESUMEN
El síndrome de Turner tiene una prevalencia de 1 en 1800 a 5000 recién nacidos vivos femeninos y se caracteriza por la ausencia total o parcial del segundo cromosoma X. Actualmente se reconoce gran variedad en la presentación citogenética, siendo la más común la monosomia del X (45,X) y entre las menos frecuentes los mosaicismos que incluyen fragmentos o la totalidad del cromosoma Y. La presencia de este cromosoma confiere características fenotípicas de androgenización y obliga a la realización de estudios de extensión en las pacientes. Se reporta un caso de síndrome de Turner con mosaicismo 45,X/46,XY, se revisa la literatura y se propone un protocolo de estudio complementario y manejo de las pacientes con este síndrome.
PALABRAS CLAVE: Síndrome de Turner, variación citogenética, fenotipo, mosaicismo
SUMMARY
Turner syndrome has an estimated prevalence of 1 in 1800 to 5000 female births and is characterized by total or partial absence of the second chromosome X. It’s now recognized that there is variation in the cytogenetic presentation of the syndrome, being the most common monosomy X (45, X) and among the least frequent it’s found mosaicism including fragments or the entire Y chromosome. The presence of Y chromosome confers phenotypic characteristics of androgenization and requires extension studies in patients. We report a case of Turner syndrome with mosaic 45,X/46,XY karyotype found by G-banding and aditionally, we propose a protocol for further studies and management of patients with this syndrome.
KEY WORDS: Turner’s syndrome, cytogenetic variation, phenotype, mosaicism
INTRODUCCIÓN
El síndrome de Turner (ST) es una condición genética caracterizada por la ausencia total o parcial del segundo cromosoma X. Su prevalencia oscila entre 1 en 1800 a 5000 recién nacidos vivos femeninos, atribuyéndose las diferencias entre prevalencias estimadas, a la población estudiada y el momento del análisis cromosómico, sin embargo, se estima que representa hasta el 3% de todas las concepciones (1).
La forma mas frecuente de presentación citogenética es 45X, sin embargo, alrededor del 50% de los pacientes presentan una formula cromosómica diferente. La variedad 45,X/46,XY es de las de menor frecuencia y esta reportada en cerca del 5% de los casos (2).
El ST tiene una expresividad variable. Se puede sospechar el diagnóstico in utero a través de la ecografía prenatal cuando se encuentra edema, hídrops fetal, higroma quístico, cardiopatías complejas, restricción de crecimiento intrauterino. En la infancia o adolescencia los hallazgos sugestivos son: talla baja, cuello alado, amenorrea primaria y signos de hipogonadismo (2-4).
En este artículo se presenta el caso de una paciente con ST cuya fórmula cromosómica era 45,X/46,XY, una de las variedades citogenéticas menos frecuentes de dicho síndrome; además presentamos una breve revisión de la literatura, el análisis clínico del caso y un protocolo de estudio y de manejo sugerido por los autores.
Caso clínico
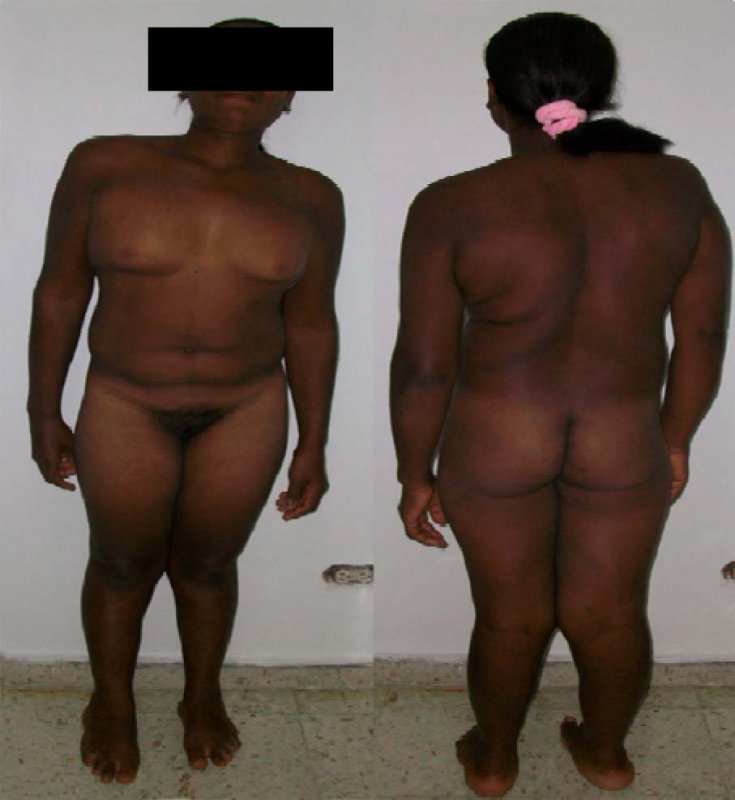
Paciente de 27 años, analfabeta, procedente del área rural de López de Micai, Cauca, Colombia. Consulta al servicio de ginecología por amenorrea primaria. Los hallazgos al examen físico fueron: talla de 145 cm (< percentil 3), distribución ginecoide del vello púbico, sin vellos en axilas y piernas, espalda amplia, musculatura masculina, manos y pies grandes (> percentil 95), cubitus valgus, genu valgus, hipoplasia del cuarto tarsiano, genitales externos femeninos de apariencia usual, con vagina corta (Figura 1). Se realizan las siguientes ayudas diagnósticas:
– Ecografía pélvica vía abdominal: ausencia de útero y ovarios.
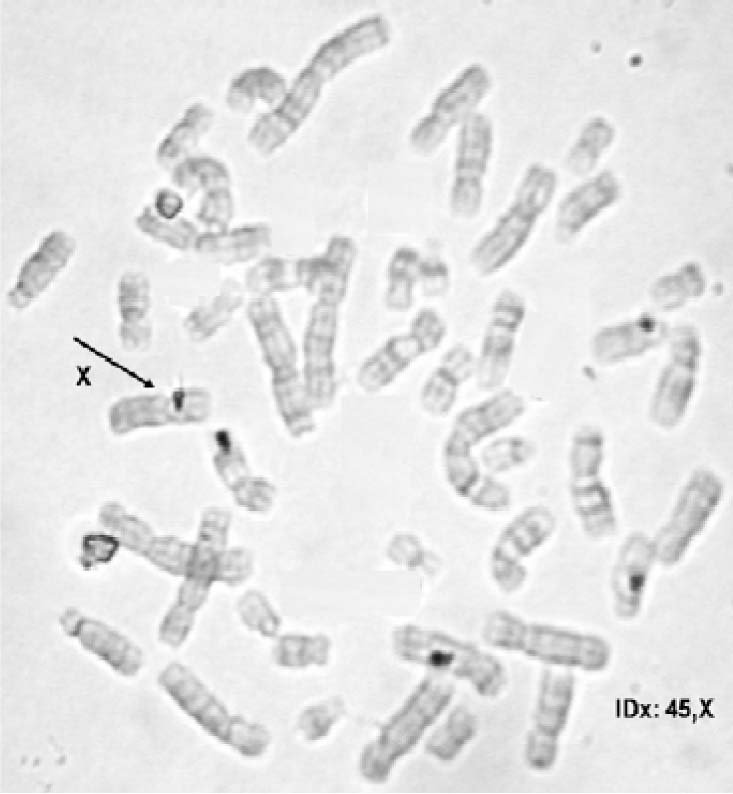
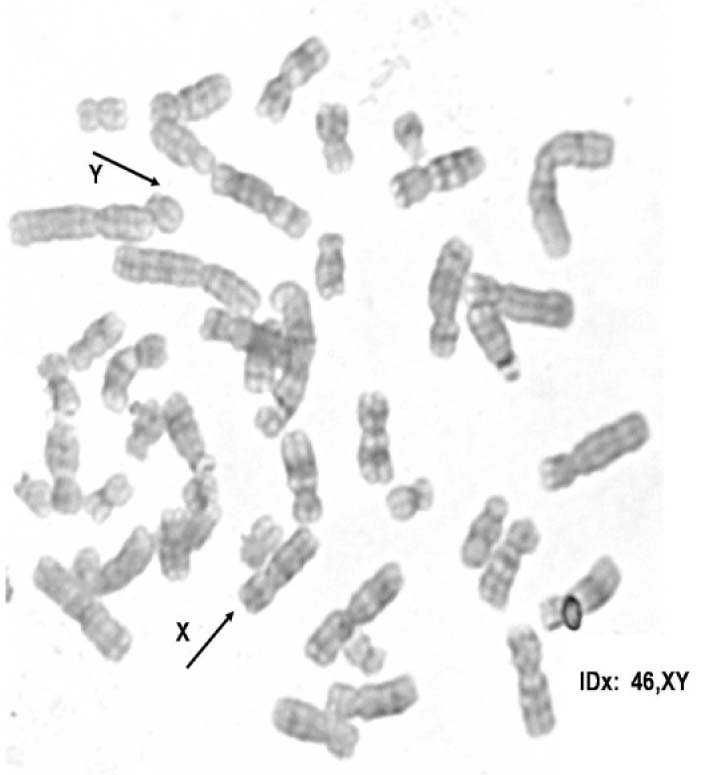
– Cariotipo con bandeo G, con resolución de 560 bandas, encuentra en 30 metafases: 45,X (Figura 2) y en 21 metafases 46,XY (Figura 3). Con estos resultados se concluyó que existía un mosaicismo 45,X/46,XY.
– Resonancia magnética nuclear (RMN) de abdomen y pelvis: ausencia de órganos genitales internos femeninos. No se evidencia estría gonadal (Figura 4).
– Estradiol: 30 pg/ml (disminuido)
– FSH: 81,8 mUI/ml (aumentada)
– Testosterona libre: 0,13 pg/ml (disminuida)
– LH: 18,6 mUI/ml (normal)
DISCUSIÓN
La frecuencia del ST en todas las concepciones es de aproximadamente el 3%, sin embargo se estima que 99% de estos fetos son abortados en forma espontánea, y que el desorden afecta solamente 1 de cada 1800 a 5000 nacidos vivos en diferentes poblaciones (1,3). La incidencia estimada del ST en la población blanca es de 25-55 casos por cada 100.000 nacidos vivos femeninos, y tanto ésta como la prevalencia aumentan al realizar estudios cromosómicos prenatales. La presencia de mosaicismos, ahora detectados por técnicas citogenéticas avanzadas, incide también sobre este aumento de la frecuencia (1,4,5). En Colombia no hay datos sobre la prevalencia ni incidencia de este síndrome.
Los pacientes con ST se caracterizan por presentar talla baja y disgenesia gonadal. Esta última alteración conlleva alteraciones endocrinológicas, presentadas por la paciente aquí reportada: 17-ßestradiol disminuido, FSH elevada, correspondiente a los niveles de una mujer menopáusica, testosterona libre en el límite inferior y niveles normales de LH, que usualmente en pacientes con ST están elevados. Los hallazgos fenotípicos más frecuentemente encontrados son: cuello ancho, implantación baja de cabello posterior, tórax ancho, hipertelorismo mamario, defectos renales y cardiovasculares, defectos müllerianos, cubitus valgus y retraso mental hasta en el 10% de pacientes. En algunos casos se presenta in utero hídrops fetal y el clásico higroma quístico (2,6,7). Estas características físicas, la mayoría encontrados en la paciente, son la base clínica para la sospecha del síndrome. Signos como manos y pies aumentados de tamaño, espalda amplia y musculatura prominente nos orientan hacia la presencia de fragmentos o la totalidad del cromosoma Y, que sugerirían un trastorno de origen cromosómico diferente al habitual como en el caso de esta paciente.
El síndrome se caracteriza por una variación importante en su presentación citogenética, siendo la más frecuente, aproximadamente 50% de los casos, la monosomía del cromosoma X. Otras variedades incluyen: 46,X,i(Xq); 45,X/46,X,i(Xq); 45,X/46,XX, 45,X/47,XXX; 45,X/46X,+anillo; 45,X/46,XY; 46,X,Xq- y 46,X,Xp- (2,4,8) (Tabla I). Existen diferencias en la frecuencia de las diferentes presentaciones citogenéticas descritas en los estudios prospectivos y series de casos de pacientes de clínicas de endocrinología y genética. Las series reportan la monosomía del X hasta en 50% de los casos, a diferencia de lo reportado por Nielsen y Wohlert (5), quienes encontraron la fórmula cromosómica 45,X presente solo en 1 de cada 9 casos de ST mientras los restantes correspondían a mosaicos y anormalidades de X.
Se considera que la detección de material correspondiente al cromosoma Y es cada vez mayor debido al uso más frecuente de técnicas como FISH, lo cual podría significar que la verdadera incidencia de este fenómeno entre pacientes con ST puede llegar a ser del 10 al 15% (4,8). En pacientes con signos de androgenización y presencia del cromosoma Y completo o fragmentos de este, confirmada con cariotipo o FISH, deben realizarse estudios imagenológicos de extensión, idealmente RMN, para descartar la presencia de estría gonadal, puesto que ésta se relaciona con el desarrollo de un raro tumor, el gonadoblastoma; lo cual obliga a su resección quirúrgica (4).
Es frecuente que las pacientes con amenorrea primaria consulten al medico ginecólogo el cual debe abordar cada caso con una historia clínica completa y un examen físico exhaustivo enfocado en la búsqueda de signos clásicos del ST, como talla baja, cardiopatías, retardo mental, alteraciones de los derivados de Müller, desarrollo anormal de las características sexuales secundarias y signos de androgenización. Entre los estudios complementarios se debe realizar el cariotipo con bandas G, de alta resolución; de mostrar una formula cromosómica que confirme el ST, se debe solicitar las siguientes ayudas diagnósticas: Estudios imagenológicos: ecografía pélvica transabdominal y/o vaginal si fuera posible, ecocardiograma y ecografía renal. Pruebas hormonales: FSH, LH, 17-ß-estradiol, testosterona libre y TSH. FISH con sondas dirigidas a fragmentos del cromosoma Y, cuando el cariotipo con bandeo G de alta resolución no mostró cromosoma Y con signos de androgenización. Otros: valoración de la audición, valoración oftalmológica, perfil lipídico, pruebas de función hepática (4).
Para el manejo de los pacientes con este síndrome, se deben seguir algunas recomendaciones sobre el uso de la terapia de reemplazo hormonal (TRH) dadas por el comité del ST reunido en Bethesda en 2007 (4,9,10):
– Inicio temprano de hormona del crecimiento, incluso desde los 9 meses de edad. – La transición puberal debe hacerse lo más fisiológicamente posible, y antes de iniciar la terapia con estrógenos, se deben determinar los niveles de gonadotropinas para excluir la posibilidad de pubertad espontánea retardada.
– Al momento de iniciar la TRH para inducir el desarrollo puberal, la forma, dosis y el tiempo deben reflejar el proceso de la pubertad normal (Tabla II).
– El inicio de esta terapia en forma temprana, no influye sobre el crecimiento que se quiere lograr a través de la terapia con hormona de crecimiento, y se ha demostrado que es benéfica para la paciente ya que logra una maduración puberal adecuada y además evita los efectos deletéreos de la falta de estrógenos sobre la densidad mineral ósea (9,10). El uso de anticonceptivos orales para obtener un desarrollo puberal debe evitarse, ya que los estrógenos sintéticos tienen dosis muy altas en la mayoría de las formulaciones y las progestinas sintéticas pueden influir en el desarrollo mamario y uterino. Es importante recordarle a la paciente que la terapia de reemplazo hormonal es necesaria hasta el tiempo normal de la menopausia, para mantener la feminización y prevenir la osteoporosis (9,10).
CONCLUSIONES
El abordaje de los pacientes con amenorrea primaria debe ser integral, incluyendo una historia clínica completa y un examen físico exhaustivo, imágenes, medición de hormonas y cariotipo con bandeo G de alta resolución. En casos de ST existen situaciones especiales en las cuales es necesario realizar FISH para detectar fragmentos del cromosoma Y, si este es el caso, se deben complementar con RNM, y resección de estría gonadal. Es necesario evaluar la necesidad del uso de hormona del crecimiento y de terapia de reemplazo hormonal para todos los casos de ST.
BIBLIOGRAFÍA
1. Lippe B. Turner syndrome. Endocrinol Metab Clin North Am 1991;20:121-52.
2. Firth H, Hurst J. Turner syndrome 45X and variants. Oxford Desk reference. Clinical genetics. 1th edition. New York. Oxford University Press 2007. Cap 5; 558-60.
3. Sybert V, McCauley E. Turner syndrome. N Engl J Med 2004;351:1227-38.
4. Gravholt C, Hjerrild B, Mortensen K. Turner syndrome and clinical treatment. British Medical Bulletin 2008;86:77-93.
5. Nielsen J, Wohlert M. Sex chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study inn Arhus, Denmark. Birth Defects Orig Artic Ser 1990;26:209-23.
6. Bondy C. Care of girls and women with Turner Syndrome. J Clin Endocrinol Metab 2007,92:10-25.
7. Davenport M. Approach to the patient with Turner syndrome. J Clin Endocrinol Metab 2010;95:1487-95.
8. Alvarez-Nava F, Soto M, Sanchez MA, Fernandez E, Lanes R. Molecular analysis in Turner syndrome. J Pediatr 2003;142:336-40.
9. Hanton L, Axelrod L, Bakalov V. The importance of estrogen replacement in young women with Turner syndrome. Womens Health (Larchmt) 2003;12:971-7.
10. Nabhan Z, DiMeglio L, Qi R, Perkins S, Eugster E. Conjugated oral versus transdermal estrogen replacement in girls with Turner syndrome: A Pilot Comparative Study. J Clin Endocrinol Metab 2009;94:2009-14.


