2017
5
Autores:
Zoraida Frías Sánchez, Amalia Rodelgo del Pino, Manuel Pantoja Garrido, José Rubio Valtueña, Julio Álvarez Bernardi
Imágen 1:
Imágen 2:
Imágen 3:
Tipo de documento:
Casos ClínicosTUMOR DE CÉLULAS DE LA GRANULOSA. NEOPLASIA ESTROMAL DE LOS CORDONES SEXUALES. REVISIÓN DE LA LITERATURA, A PROPÓSITO DE DOS CASOS CLÍNICOS.
Contenido del documento:
RESUMEN
Los tumores de las células de la granulosa, hacen referencia a un conjunto de neoplasias derivadas del tejido estromal de los cordones sexuales, secretores de estrógenos, que corresponden al 1-5% de todos los tumores malignos del ovario, aunque pueden presentar localizaciones extraováricas. Son tumores raros e infrecuentes, cuya incidencia general varía de 0,4 a 1,7 casos por cada 100.000 mujeres al año. Se clasifican en dos grupos diferenciados en función de la edad de las pacientes, su historia natural y sus características patogénicas: tumores de células de la granulosa tipo adulto (TCGA) o tipo juvenil (TCGJ). El diagnóstico de esta rara patología se basa en cuatro pilares fundamentales, por un lado, las manifestaciones clínicas y la confirmación mediante pruebas de imagen, y, por otro lado, los marcadores tumorales y el estudio inmunohistoquímico de las muestras biológicas. En el manejo terapéutico de los estadios iniciales de la enfermedad (supervivencia a los 5 años superior al 90% en los estadios I), se recomienda el tratamiento quirúrgico mediante histerectomía con anexectomía bilateral en pacientes con TCGA y deseos genésicos cumplidos, reservando la anexectomía unilateral para los casos de TCGJ o para aquellas pacientes sin descendencia (aunque con posterioridad se recomienda completar el tratamiento). La quimioterapia sistémica postoperatoria se incluye cuando existe extensión extraovárica o recurrencia de la enfermedad. La tasa de recidiva (considerado uno de los principales factores de mal pronóstico), es alta en estadios avanzados, aunque la probabilidad de desarrollar metástasis a distancia es baja.
PALABRAS CLAVE: Tumor células granulosa, neoplasias ovario, tumores estromales cordones sexuales, neoplasias genitales.
SUMMARY
Tumors of the granulosa cells refer to a set of neoplasms derived from the stromal tissue of the sex cords, secretory of estrogens. They correspond to 1-5% of all malignancies of the ovary, though they may have extraovarian locations. These tumors are rare and uncommon, whose general incidence may vary from 0.4 to 1.7 cases per 100,000 women per year. They are classified into two groups depending on the age of the patients, their natural history and their pathogenic characteristics: adult type tumors of the granulosa cell (AGCT) or juvenile type (JGCT). The diagnosis of this rare pathology is based on the study of clinical manifestations and confirmation through image tests, and also on tumor markers tests and the immunohistochemical study of biological samples. In the therapeutic handling of the early stages of the disease (5-year survival greater than 90% in stage I), surgical treatment via hysterectomy with bilateral adnexectomy in patients with AGCT and met genesic perspectives is recommended; for the cases of JGCT or for those patients without progeny, preserving the unilateral adnexectomy is justified (although completion of the treatment is recommended at later stages). Postoperative systemic chemotherapy is practiced when there is extraovarian extension or recurrence of the disease. The rate of relapse (considered one of the main factors of poor prognosis), is high in advanced stages, although the probability of developing distant metastasis is low.
KEY WORDS: Granulosa cell tumors, ovarian neoplasms, sex cord-gonadal stromal tumors, genital neoplasms.
INTRODUCCIÓN
Los tumores de las células de la granulosa (TCG) agrupan un conjunto de neoplasias derivadas de los tejidos estromales de los cordones sexuales1. Fueron descritos por primera vez por Rokitasnky en el año 18591, y se caracterizan por su bajo potencial de malignidad, recurrencias tardías y escasa tasa de metástasis a distancia2. Los tumores de la granulosa representan en torno al 2-6% de las neoplasias ováricas (hay que tener en cuenta que el cáncer de ovario es la tercera neoplasia del tracto genital femenino más frecuente en la mujer3), y casi el 70% de los tumores estromales de los cordones sexuales1,3. Son tumores con una gran actividad hormonal derivada de la afectación sobre las células tecales luteinizantes, produciendo una elevación de los niveles de estradiol en sangre3. El diagnóstico se basa en la asociación de manifestaciones clínicas tales como distensión abdominal, pubertad precoz, virilización o sangrado uterino anormal asociado a hiperestrogenismo/hiperplasia endometrial4, y la posterior confirmación mediante pruebas de imagen, como la tomografía axial computerizada (TAC) o resonancia nuclear magnética (RNM), aunque la ecografía, sigue siendo la técnica de exploración por imagen más usada y que mejores resultados coste/efectividad proporciona5. El tratamiento de elección en este tipo de neoplasias, es la resección quirúrgica completa del tumor, asociada a tratamientos quimioterápicos u hormonales adyuvantes (aunque sólo la quimioterapia sistémica, parece obtener una respuesta considerable, aunque esta sea parcial (tasa de remisión en torno al 50%))6. La tasa de recurrencias (factor de mal pronóstico de gran importancia), de estos tumores se encuentra entre el 9-35%, desarrollándose en la mayoría de los casos en los 10 primeros años tras el diagnóstico, con una baja probabilidad de desarrollar metástasis a distancia (5-6 %)2.
A continuación, presentamos dos casos clínicos de pacientes diagnosticadas de tumores de células de la granulosa mediante pruebas de imagen y tratadas quirúrgicamente. En el primer caso destaca que la tumoración se desarrolló a nivel retroperitoneal, en una paciente que fue intervenida de histerectomía total más doble anexectomía por útero polimiomatoso, 28 años antes. En el segundo caso, el diagnóstico se obtuvo tras una anexectomía bilateral laparoscópica por masa pélvica a filiar, en una paciente histerectomizada 5 años antes por episodios de metrorragia postmenopáusica. Nuestro objetivo es reportar los escasos casos de tumores de células de la granulosa diagnosticados en nuestro centro. Aportando a la literatura científica información para el manejo de los mismos, a través de una exhaustiva revisión de la literatura más reciente sobre el tema.
CASOS CLÍNICOS
Caso 1
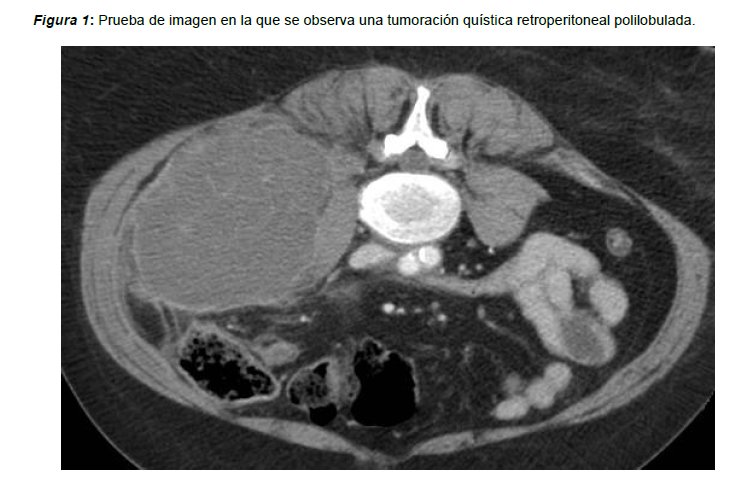
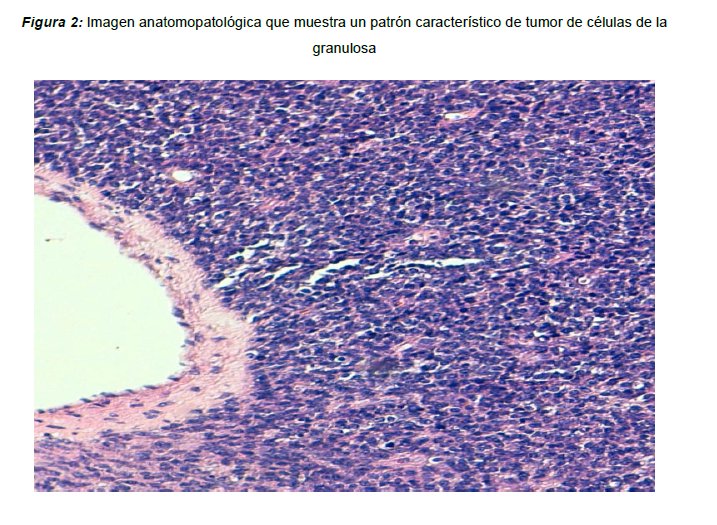
Exponemos el caso de una mujer de 68 años de edad que presenta como antecedentes personales de interés un quiste hidatídico hepático y episodios recurrentes de litiasis renal. La paciente es tercigesta, con antecedentes de dos partos eutócicos y un aborto, y fue intervenida a los 40 años de edad de una histerectomía total más doble anexectomía por útero polimiomatoso. Acude a consultas de medicina general por dolor a nivel de fosa renal izquierda, resistente a tratamiento analgésico, por lo que se solicita resonancia nuclear magnética (RNM) abdominopélvica. Los resultados indican la presencia de una tumoración quística retroperitoneal polilobulada de difícil delimitación (Figura 1), compatible con recidiva peritoneal de hidatidosis, acompañado de adenopatías inespecíficas a nivel periaórtico. Se solicita serología que resulta negativa para hidatidosis. Se discute el caso en el comité multidisciplinar de tumores, indicando extirpación quirúrgica de la tumoración retroperitoneal y adenopatías periaórticas, asociado a tratamiento coadyuvante con Albendazol en función de los resultados anatomopatológicos. La intervención quirúrgica trascurre sin incidencias completando la cirugía programada por el comité. Los resultados histológicos y anatomopatológicos muestran hallazgos morfológicos junto a patrón de expresión inmunohistoquímico, compatibles con tumor de células de la granulosa tipo adulto (Figura 2) con ganglio linfático periaórtico infiltrado por continuidad por la neoplasia. El estudio inmunohistoquímico presenta positividad para Alfa-inhibina en más del 75% de las células, con receptores de Estrógenos/Progesterona positivos en el 95% de las células estudiadas. La evolución postoperatoria de la paciente es favorable, recibiendo el informe de alta domiciliaria con seguimiento en consultas externas, tras rechazar el tratamiento quimioterápico adyuvante. El seguimiento posterior por parte de la unidad de oncología médica se encuentra dentro de la normalidad, hasta que 6 años después de la intervención, en la tomografía axial computerizada (TAC) de control, se objetiva el crecimiento de una adenopatía a nivel de la vena iliaca externa izquierda de 25 mm de eje corto (presentaba 9 mm en el TAC previo), de aspecto patológico. Por otro lado, se observa una adenopatía de 11 mm a nivel de la vena renal izquierda, sin variaciones con respecto al TAC de control previo. La exploración ginecológica se encuentra dentro de la normalidad. Se discute el caso en el comité de tumores ginecológicos, indicándose una exploración de la cavidad abdominal y resección de las adenopatías descritas, si fuera técnicamente posible. La paciente es sometida a una laparotomía exploradora, observándose un nódulo pélvico intensamente adherido a vena ilíaca externa izquierda, sospechoso de recidiva de tumor de células de la Granulosa. Se procede a la exéresis de dicho nódulo, además de la resección de una muestra de peritoneo parietal y de una adenopatía sospechosa al tacto, a nivel de la bifurcación iliaca. El servicio de cirugía general realizó una exéresis del saco herniario asociado a la cirugía previa, y de un implante a nivel de intestino delgado. Las piezas quirúrgicas son remitidas al servicio de Anatomía Patológica, obteniendo como resultado, negatividad para malignidad de todas las muestras excepto del nódulo iliaco externo izquierdo, donde se objetiva un patrón histológico compatible con recidiva de tumor de células de la Granulosa tipo adulto. Se decide posteriormente derivar a la paciente a la unidad de Oncología Médica para iniciar tratamiento con quimioterapia sistémica, pero la paciente lo rechaza, aduciendo no desear más seguimiento ni tratamiento para su cuadro clínico.
Caso 2
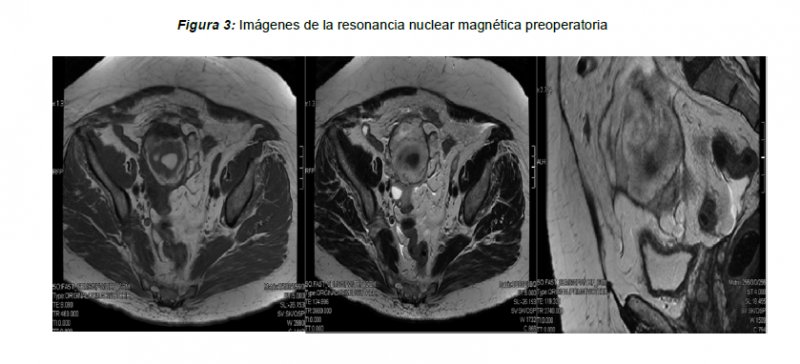
Exponemos el caso de una mujer de 73 años de edad que presenta como antecedentes personales de interés un pseudotumor intracraneal tratado mediante derivación lumboperitoneal hace 13 años, espondilosis, múltiples hernias discales, hipertensión intracraneal benigna y gastritis crónica. La paciente es quintigesta, con antecedentes de cuatro partos eutócicos y un aborto, con menopausia a los 45 años de edad. Como antecedentes quirúrgicos destacan una colecistectomía y una apendicectomía. La paciente acude a las consultas de ginecología general tras varios episodios de metrorragia postmenopáusica. La exploración ginecológica es normal, observándose en la ecografía ginecológica un útero atrófico con endometrio engrosado de 15 mm, no visualizándose los anejos. Se realiza una citología cervical que es informada como negativa para malignidad. Se deriva a la paciente a la consulta de histeroscopia, observándose durante la misma una cavidad endometrial de aspecto proliferativo con múltiples focos hemorrágicos, por lo que se procede a tomar varias biopsias dirigidas. El resultado histológico de las muestras objetiva la presencia de un endometrio normal, débilmente proliferativo. Ante la persistencia de la sintomatología se propone la realización de una histerectomía total vía vaginal, que la paciente acepta. La intervención discurre sin complicaciones, recibiendo la paciente el alta hospitalaria, tras una evolución postoperatoria favorable. El estudio anatomopatológico fue normal, observándose una adenomiosis uterina asociada a endometrio proliferativo y cérvix con metaplasia escamosa. Cinco años después, en el contexto de un estudio de extensión por episodios de síncope asociado a dolor abdominal inespecífico, se observa en el TAC abdominopélvico, una imagen compatible con tumoración en fosa iliaca derecha (FID) de aproximadamente 7,8×8,7×8,7 cm, hiperdensa que contacta con otra imagen de mayor atenuación y de menor tamaño (2,8×6,2×3,3 cm), que se encuentra en contacto con la pared anterior del sigma, compatible con hematoma mesentérico, sin otros hallazgos de interés. La paciente es ingresada para valoración multidisciplinar, sufriendo una recaída en su cuadro clínico, con aumento de las algias abdominales, por lo que se solicita interconsulta al servicio de ginecología y resonancia nuclear magnética (RNM) abdominopélvica (Figura 3). La prueba de imagen indica la presencia de una formación nodular en región anexial derecha de 2,8×3,6×2,8 cm, sugestiva de corresponder al anejo derecho aumentado de tamaño. Anterior a dicho anejo, se observa formación heterogénea de contornos mal definidos de aproximadamente 8×7,6×8 cm, compatible con hematoma evolucionado, y escasa cantidad de líquido libre en pelvis. Anterior a los vasos ilíacos externos del lado izquierdo, se aprecia imagen fusiforme de 2×1.3 cm relacionada con anejo izquierdo. Estas imágenes sugieren una alta probabilidad de actividad ovárica. La exploración ginecológica se encuentra dentro de la normalidad, objetivándose en la ecografía transvaginal una imagen compatible con anejo derecho, en el que se aprecian varias formaciones foliculares (la mayor de 15 mm), adyacente a otra formación heterogénea sugestiva de hematoma organizado de 84×54 mm. La paciente es derivada a consultas externas de ginecología tras recibir el alta hospitalaria. Se solicitan marcadores tumorales, que se encuentran dentro de la normalidad, excepto por una elevación del estradiol (83 pg/ml) y de la inhibina B (622 pg/ml). Ante los resultados de las pruebas anteriores, se decide realizar una laparoscopia exploradora más anexectomía bilateral y citología de lavado peritoneal. La intervención quirúrgica transcurre dentro de lo previsto, recibiendo la paciente el alta hospitalaria, tras una evolución favorable. Los resultados anatomopatológicos de las muestras del líquido peritoneal y del anejo izquierdo se encentran dentro de la normalidad. Sin embargo, el estudio histopatológico del anejo derecho refleja la presencia de cuerpos de Call Exner (Figura 4) y células de pequeño tamaño, sin atipia y con pleomorfimo, característico de un tumor de células de la Granulosa del adulto (estadio IA de la FIGO (Federación Internacional de Ginecología y Obstetricia)). La paciente presenta una evolución postoperatoria muy favorable, recibiendo el alta hospitalaria a los 2 días de la intervención. Actualmente continúa en seguimiento por las unidades de oncología médica y ginecología de nuestro centro. No ha presentado recurrencias de la enfermedad, y tanto las pruebas de imagen como los niveles de Inhibina A y B seriados periódicamente, se encuentran dentro de la normalidad.
DISCUSIÓN
El nombre de tumores de células de la granulosa fue propuesto por von Werdt en 19147 y hace referencia a un conjunto de neoplasias derivadas del tejido estromal de los cordones sexuales, secretores de estrógenos4, que corresponden al 1-5% de todos los tumores malignos del ovario4,7-8, aunque pueden presentar localizaciones extraováricas7 (por ejemplo, se han divulgado sólo 12 referencias bibliográficas de formas retroperitoneales en la literatura médica en inglés, desde 1938 hasta nuestros días7). Son tumores raros e infrecuentes, cuya incidencia general varía de 0,4 a 1,7 casos por cada 100.000 mujeres al año1,4. Se clasifican en dos grupos diferenciados en función de la edad de las pacientes, su historia natural y sus características patogénicas: tumores de células de la granulosa tipo adulto (TCGA) o tipo juvenil (TCGJ), definida por primera vez por Scully en el año 19771,4,8. Los TCGA son la formas más frecuentes (95%) y aparecen normalmente a partir de los 40 años de edad, con un pico de incidencia entre los 50-55 años, mientras que las formas juveniles son más raras (5%), presentándose preferentemente en torno a los 20 años de edad, o incluso antes (45% en la premenarquia)1,4,8. Nuestras pacientes por lo tanto se clasificarían dentro del primer grupo. Desde el punto de vista histológico, estas neoplasias se pueden dividir en pobremente diferenciadas o bien diferenciadas, estas últimas se caracterizan por presentar un patrón celular folicular, trabecular y tubular, observándose en un 30% de los casos cuerpos de Call-Exner1. Los cuerpos de Call-Exner se definen como acúmulos de células de la granulosa en forma de pequeños anillos, rodeados por un material eosinófilo y núcleos disminuidos de tamaño1. Por otro lado, los pobremente diferenciados, se caracterizan por presentar un patrón más difuso, con un estroma esclerótico secundario a cambios degenerativos y procesos isquémicos1. Los TCGA ováricos son neoplasias hormonalmente activas que se caracterizan por su capacidad para expresar aromatasa y secretar esteroides sexuales, mientras que las formas extraováricas derivan de tejido estromal gonadal ectópico migrado a través del mesonefro7. Independientemente de su origen, estas células presentan un citoplasma monomórfico redondeado con núcleos ovales y cromatina granular7. Nuestra primera paciente parece haber desarrollado una tumoración retroperitoneal extraovárica, por ello no presenta la positividad para marcadores como el estradiol, que si presenta la segunda (cuyo origen ovárico parece más claro). Etiológicamente, los tumores de células de la granulosa parecen estar relacionados con la exposición continua a drogas inductoras de la ovulación, anomalías cromosómicas como la trisomía 12, monosomía 22 y delección de la rama larga del cromosoma 6, enfermedades y síndromes como Peutz-Jeghers, Potters, Ollier o Maffucci1. Además, las alteraciones en el gen FOXL2, puede tener un papel conductor en la patogénesis del TCGA9, mientras que la vía Hipona/YAP puede influir en la regulación de la proliferación/migración celular y la esteroidogénesis9. Las mutaciones específicas del FOXL2 en el punto C134W están presentes en el 90% de los tumores de células de la granulosa ováricos, ya que desempeña un papel importante en el incremento de la actividad de la aromatasa, que promueve y favorece el desarrollo y progresión de este tipo de neoplasias9-10. El diagnóstico de esta rara patología se basa en cuatro pilares fundamentales, por un lado, las manifestaciones clínicas y la confirmación mediante pruebas de imagen, y por otro lado, los marcadores tumorales y el estudio inmunohistoquímico de las muestras biológicas. Clínicamente los TCG se caracterizan por desarrollar síntomas inespecíficos similares a los de otras masas pélvicas como distensión abdominal, tumoración palpable, dolor, rotura/torsión de masas anexiales, etc…1,3,8,11-12. Estos tumores producen trastornos hormonales asociados a la liberación de hormona folículo estimulante (FSH), estradiol o prolactina, desencadenando cuadros de oligo/amenorrea, trastornos menstruales, metrorragia postmenopáusica, hiperplasia endometrial, adenocarcinoma de endometrio, síndrome de hiperestimulación ovárica, hiperprolactinemia o galactorrea1,3,8,11-12. Dentro de estas manifestaciones, tienen gran importancia la estimulación hormonal sobre el endometrio, que deriva en que casi el 10% de las pacientes con TCG se asocien a adenocarcinomas de endometrio, por lo que es recomendable en estas pacientes, la realización de una biopsia endometrial8. Los TCGJ están asociados además a otros cuadros clínicos relacionados con la pubertad precoz, esclerosis tuberosa, síndromes congénitos como la enfermedad de Ollier, síndrome de Maffuci, Goldenher o Potter, entre otros1,3,8,11. En el primer caso, el dolor abdominal compresivo por la masa retroperitoneal fue el síntoma inicial, sin embargo en el segundo caso, si se presentaron manifestaciones derivadas del hiperestrogenismo característico de este tipo de tumores, como fueron los sangrados postmenopáusicos secundarios al engrosamiento endometrial (ninguna de las pacientes presentaba un trofismo genital anormal para su edad). Tras la sospecha clínica, las pruebas de imagen permiten una mayor aproximación hacia un diagnóstico definitivo, siendo la ecografía y el TAC, las técnicas más usadas. Los patrones radiológicos de estas masas son difíciles de diferenciar de los de otras tumoraciones anexiales o pélvicas, variando desde las formas solidas heterogéneas u homogéneas, hasta otras uniloculares o multiloculares con diferentes grosores a nivel parietal1. Los TCG suelen desarrollarse como masas sólidoquísticas, unilaterales, polilobuladas, con regiones hemorrágicas intratumorales necróticas en áreas centrales, degeneración fibrosa, calcificaciones, tabiques, estructuras papilares y una apariencia sólida heterogénea de gran tamaño (entre 10-15 cm de diámetro (rango 1-30cm)), lo que dificulta el diagnóstico diferencial con los cistoadenomas, las neoplasias ováricas o los sarcomas uterinos1,8. La RNM también puede tener un papel importante en el diagnóstico de los TCG, aunque no parece que aporte ningún beneficio extra a las otras técnicas descritas anteriormente8. En nuestros casos, ambas pacientes fueron sometidas a diferentes pruebas radiológicas (ecografía, TAC y RNM). Por otro lado, tanto los marcadores tumorales como el estudio inmunohistoquímico permiten aportar un dato de confirmación para reforzar el diagnóstico de los TCG. Los elementos inmunohistoquímicos más utilizados en el estudio de estas neoplasias son la inhibina-A, vimetina, AE1/AE3, CD56, Ki-67, p53 y el CD99, mientras que, desde el punto de vista de los marcadores tumorales en sangre, los más importantes son la inhibina B, hormona antimülleriana (AMH), proteína reguladora del folículo (FRP) y el 17 β Estradiol 1,13. Un Ki-67 superior al 60% está relacionado con tumores de curso más agresivo, mientras que la positividad para inhibina-A, vimetina, p53 o CD56, está asociado a mayor componente sarcomatoide de la masa13. La positividad para inhibina-B y para AMH puede estar relacionado, durante el seguimiento posterapéutico, con la posibilidad de recidiva tumoral1. La primera de nuestras pacientes presentó un estudio inmunohistoquímico altamente positivo para Alfa-inhibina con receptores de estrógenos/progesterona positivos; mientras que los marcadores tumorales en el caso de nuestra segunda paciente fueron normales, excepto por el estradiol y la inhibina-B, que estaban ampliamente elevados (sobre todo el segundo). El manejo terapéutico de los TCG es muy controvertido actualmente, sobre todo el referente a la enfermedad recurrente o metastásica1,2,6,10,14. Parece que sí existe consenso en el manejo de los estadios iniciales de la enfermedad, donde se recomienda el tratamiento quirúrgico mediante histerectomía con anexectomía bilateral en pacientes con TCGA y deseos genésicos cumplidos, reservando la anexectomía unilateral para los casos de TCGJ o para aquellas pacientes sin descendencia (considerando completar la resección quirúrgica cuando los deseos genésicos se cumplan, debido a la alta tasa de recurrencias)1. En los casos de enfermedad avanzada o metastásica, es preciso la realización de resecciones quirúrgicas agresivas asociadas a quimioterapia sistémica postoperatoria, ya que esta parece mejorar la supervivencia a largo plazo de estas pacientes2. El papel de la linfadenectomía también es discutible, ya que estudios como los de Ayhan et al indican que la presencia de ganglios linfáticos metastásicos son infrecuentes14-15. La utilización de otras estrategias terapéuticas como la quimioterapia intraperitoneal hipertérmica, los tratamientos con inhibidores de la aromatasa o la radioterapia, no parecen estar lo suficientemente apoyados aún, por estudios que demuestren su eficacia a largo plazo, aunque pueden tener algún futuro en el control de la enfermedad residual o recurrente2,6,10. En nuestras dos pacientes, el tratamiento indicado se basó en la extirpación quirúrgica de las lesiones sospechosas. Mientras que, en el primer caso, el estadio se consideró avanzado, recomendándose el inicio de tratamiento mediante quimioterapia sistémica (que la paciente rechazó); en el segundo caso, el estadio fue tan inicial y por lo tanto de buen pronóstico, que se optó por el seguimiento anual en consultas. Se considera que el diagnóstico y tratamiento de los TCG en estadios iniciales, es uno de los principales factores pronósticos positivos de esta enfermedad, ya que la tasa de supervivencia a los 5 años es superior al 90% en los estadios I de la FIGO, reduciéndose hasta un 25-45% en los estadios II/III1,2,11. Otros factores de buen pronóstico de gran importancia, son los referentes a la edad inferior a 40 años, tamaño tumoral menor de 10 cm, ausencia de rotura tumoral, recurrencias o enfermedad residual, actividad mitótica atípica menor de 5/10 mitosis por campo (HPF), ausencia de aneuploidías o sobrexpresión de la p53 e índice Ki-67 bajo1. Los TCG son neoplasias de crecimiento lento, pero que poseen una tasa de recurrencia alta (las series más optimistas, indican una probabilidad de recurrencia del 20% como mínimo), que se considera uno de los principales factores de mal pronóstico, ya que aumenta la tasa de mortalidad hasta valores cercanos al 70-80%1,9. El tiempo medio de aparición de recidivas se encuentra en torno a los 75 meses (rango de 6-166), aunque se han reportado casos de recurrencia hasta 40 años después del tratamiento1. Por otro lado, la difusión metastásica de estos tumores es vía hematógena (la probabilidad de metástasis linfáticas es inferior al 10%8), siendo muy rara en estadios iniciales y pudiendo afectar a cualquier órgano, aunque se producen más frecuentemente en hígado, pulmón y cerebro8-9. La supervivencia en 5 años de los pacientes con metástasis extraováricas de TCG, se reduce hasta valores de un 33-50%9. Nuestro primer caso nos lleva a pensar que en un TCGA de origen retroperitoneal (con ganglio linfático periaórtico infiltrado por continuidad), ya que la paciente fue sometida a una histerectomía total más doble anexectomía 28 años antes y, aunque se han reportado recidivas tumorales hasta 40 años después, no parece el caso. Sin embargo, la paciente sufrió una recurrencia de su enfermedad a los 6 años de la intervención, y a nivel linfático, que se considera una localización muy infrecuente de metástasis, en este tipo de tumores. En el segundo caso, la paciente fue intervenida de una histerectomía total simple vía vaginal, por episodios repetidos de metrorragia postmenopáusica con endometrio engrosado débilmente proliferativo, confirmado por el servicio de Anatomía Patológica. Pero a los 5 años de la cirugía, se observó la presencia de una lesión nodular anexial derecha que tras exéresis laparoscópica, resulto corresponder a un TCGA, lo que nos puede llevar a pensar en la posibilidad de que la afectación neoplásica ya estuviera presente microscópicamente, cuando la paciente refirió los síntomas hemorrágicos, ya que el engrosamiento endometrial y el sangrado, son dos manifestaciones clínicas típicas de este tipo de tumores, derivada de la estimulación estrogénica, confirmada analíticamente a posteriori (Estradiol elevado en sangre). Debemos enfatizar que en ambos casos el diagnóstico de TCGA se produjo en pacientes con histerectomías previas por otra causa, haciendo aún más especiales estos dos reportes. En la literatura observamos publicaciones similares, aunque muy escasas, de pacientes histerectomizadas entre 10-22 años antes del diagnóstico de TCGA, por úteros miomatosos en su mayoría7,16-17. La localización de dichas neoplasias es muy heterogénea (desde anexial hasta retroperitoneal o mesentérica)7,16-17. Parece existir una relación causal para los casos anteriormente descritos, ya que los miomas son tumores estrógeno dependientes y los TCG son neoplasias productoras de estradiol, derivadas de la afectación sobre las células tecales luteinizantes16. Por otro lado, tanto el desarrollo primario como las recidivas retroperitoneales de este tipo de tumores son también infrecuentes, teniendo en cuenta que casi un 52% de las mismas se producen en localizaciones extraováricas18. Lo más importante en el manejo de los TCG retroperitoneales, es tener en cuenta que no existe consenso en las terapias adyuvantes necesarias para su control, pero que el tratamiento hormonal puede jugar un papel preponderante dentro del tratamiento.7,18-19. Además, es necesario un exhaustivo diagnóstico diferencial con otro tipo de neoplasias que se pueden desarrollar en estos territorios anatómicos, como el carcinoma de células pequeñas, carcinoma indiferenciado, los tumores carcinoides o los linfomas, pudiendo diferenciarlos entre otros aspectos, por las pruebas inmunohistoquímicas7.
CONCLUSIONES
Los TCG son neoplasias derivadas del estroma de los cordones sexuales, que corresponden a menos del 5% de la totalidad de cánceres de ovario, diagnosticados en las mujeres. Se caracterizan clínicamente por sintomatologías relacionadas con la elevación sanguínea de determinadas hormonas, cuya producción estimulan. El diagnóstico se apoya tanto en las pruebas de imagen, como en el estudio inmunohistoquímico y de una serie de marcadores tumorales, característicos de este tipo de patologías. Las terapias utilizadas son controvertidas, ya que no existen grandes protocolos consensuados sobre el manejo de los TCG, aunque parece de elección la resección quirúrgica de la tumoración en estadios iniciales, asociado a quimioterapia sistémica cuando existe extensión extraovárica. La tasa de recurrencias es elevada en aquellas pacientes con factores de mal pronóstico, lo que produce un aumento considerable de la mortalidad y una reducción de la supervivencia a los 5 años de entorno al 30-50%. Por otro lado, tenemos que tener en cuenta que tanto la localización retroperitoneal de la masa, como la historia previa de histerectomía (con o sin doble anexectomía), no son datos que nos deban hacer descartar a esta rara patología dentro del diagnóstico diferencial de las neoplasias ginecológicas.
BIBLIOGRAFÍA
1. Núñez Troconis J, Vilora ME, Delgado M and Romero R. Tumor de las células de la granulosa: importancia de la inmunohistoquímica en el diagnóstico. Rev Obstet Ginecol Venez 2016;76(2):143-149.
2. Fumihiko Fujita et al. A recurrent granulosa cell tumor of the ovary 25 years after the initial diagnosis: A case report. Int J Surg Case Rep. 2015; 12: 7–10.
3. Anuradha C.K. Rao, Manjari Kishore and Vidya Monappa. Juvenile Granulosa Cell Tumour: Anaplastic Variant with Omental Deposits. J Clin Diagn Res. 2016 Feb; 10(2): ED01–ED03.
4. Anandpara KM et al. Juvenile Granulosa Cell Tumour of the Ovary with Unilocular Pure Cystic Presentation: A Case Report and Review of Literature Pol J Radiol. 2016; 81: 120–124.
5. Azahouani A and Balahcen M. Juvenile granulosa tumor of the ovary: report of a case. Pan Afr Med J. 2015; 21: 114.
6. Dogan A, Solass W and Tempfera CB. Cytoreductive surgery followed by hyperthermic intraperitoneal chemotherapy for recurrent adult granulosa cell tumor: A case report. Gynecol Oncol Rep. 2016 Apr; 16: 21–23.
7. Priya Poickattusseril Vasu, Jayalakshmy Payippat Leelamma, Babitha Alingal Mohammed and Jyotsna Yesodharan. Primary granulosa cell tumor of retroperitoneal origin: A rare presentation with emphasis on cytomorphology. J Cytol. 2016 Jan-Mar; 33(1): 52–54.
8. Rusterholz KR and MacDonald W. An Unusual Case of Juvenile Granulosa Cell Tumor of the Ovary. Radiol Case Rep. 2009; 4(1): 178.
9. Hua G et al. The four and a half LIM domains 2 (FHL2) regulates ovarian granulosa cell tumor progression via controlling AKT1 transcription. Cell Death Dis. 2016 Jul; 7(7): e2297
10. Schwartz M and Huang GS. Retreatment with aromatase inhibitor therapy in the management of granulosa cell tumor. Gynecol Oncol Rep. 2016 Jan; 15: 20–21.
11. Iqbal A, Novodvorsky P, Lubina-Solomon A, Kew FM and Webster J. Juvenile granulosa cell tumour of the ovary presenting with hyperprolactinaemic amenorrhoea and galactorrhoea. Endocrinol Diabetes Metab Case Rep. 2016; 2016: 160006.
12. Ali Ghribi, Aicha Bouden, Manef Gasmi and Mourad Hamzaoui. Unusual malignant neoplasms of ovary in children: two cases report. Korean J Pediatr. 2016 Nov; 59(Suppl 1): S107–S111.
13. Sonoyama A et al. Aggressive Granulosa Cell Tumor of the Ovary with Rapid Recurrence: a Case Report and Review of the Literature. Kobe J. Med. Sci., Vol. 61, No. 4, pp. E109-E114, 2015.
14. Mei-Leng Cheong, Jenta Shen, Shih-Hung Huang and Tsai-Yen Chien. Long-term survival in a patient with an advanced ovarian juvenile granulosa cell tumor with para-aortic lymph node metastasis. Taiwanese Journal of Obstetrics & Gynecology 55 (2016) 907e909.
15. Ayhan A, Tuncer ZS, Tuncer R, Mercan R, Yüce K, Ayhan A. Granulosa cell tumor of the ovary. A clinicopathological evaluation of 60 cases. Eur J Gynaecol Oncol 1994; 15:320e4.
16.Mohana Vamsy Chalanki et al. Granulosa cell tumor induced massive recurrence of post hysterectomy leiomyoma. Indian J Nucl Med. 2014 Jul-Sep; 29(3): 179–181.
17.Manjiri R. Naniwadekar and N. J. Patil. Extraovarian Granulosa Cell Tumor of Mesentery: A Case Report. Patholog Res Int. 2010; 2010: 292606.
18. Zani S, Stoecker M, Cox MW, Alvarez Secord A and Blazer DG. Recurrent granulosa cell tumor presenting with spontaneous retroperitoneal hemorrhage: A case report. Gynecol Oncol Case Rep. 2011 Dec; 1(1): 14–16.
19. Teoh D, Freedman R and Soliman PT. Nearly 30 Years of Treatment for Recurrent Granulosa Cell Tumor of the Ovary: A Case Report and Review of the Literature. Case Rep Oncol. 2010 Jan-Apr; 3(1): 14–18.


