2018
3
Autores:
Albaro Nieto, Gustavo Munera, Harry Pachajoa, Joaquín Rosales, María Fernanda Escobar
Imágen 1:
Imágen 2:
Tipo de documento:
Casos ClínicosENFERMEDAD DE GAUCHER, DIAGNOSTICO EN EL EMBARAZO, REPORTE DE CASO Y CONSIDERACIONES DE SU TRATAMIENTO EN COLOMBIA.
Contenido del documento:
RESUMEN
Introducción y Objetivo: La enfermedad de Gaucher es una condición autosómica recesiva de baja prevalencia, de difícil diagnóstico y manejo, especialmente en embarazo. Reportamos una gestante con enfermedad de Gaucher manejada desde la semana 15,3 hasta el término del embarazo con Taliglucerasa en la Unidad de Alta Complejidad Obstétrica, en la Fundación Valle del Lili en Cali, Colombia.
Métodos: Reporte de caso de gestante con diagnóstico de enfermedad de Gaucher diagnosticada durante el embarazo, con exacerbación de síntomas, quien presento severa pancitopenia y esplenomegalia.
Resultados: El manejo medico interdiscilplinario permitió el control del severo deterioro clínico de la paciente durante el parto, presentó hemorragia postparto con choque hipovolémico, con adecuada respuesta al manejo médico. La madre y el neonato fueron dados de alta sin otras complicaciones asociadas.
Conclusión: El manejo interdisciplinario es indispensable en gestantes con esta entidad para lograr buenos resultados maternos y perinatales.
| Abreviaturas | Significados |
|---|---|
| EG | Enfermedad de Gaucher |
| GBA1 | Enzima beta-glucosidada ácida |
| GC | Glucosil-ceramida |
| HPP | Hemorragia PostParto |
| TRE | Terapia de Reemplazo Enzimático |
| TRS | Terapia de Reducción de Substrato |
PALABRAS CLAVE: Enfermedad de Gaucher, embarazo, trombocitopenia, deficiencia beta glucocerebrosidasa acida, depósito lisosomal, hepatomegalia, esplenomegalia.
SUMMARY
Introduction and Objective: Gaucher disease is a low prevalence autosomal recessive condition, difficult to diagnose and manage especially during pregnancy. We reported a pregnant woman with Gaucher disease managed with Taliglucerase in a critical care obstetric unit from week 15.3 until the end of her pregnancy, at the Fundación Valle del Lili, Cali, Colombia.
Methods: A case report of a pregnant woman diagnosed during her pregnancy with Gaucher disease, who presented severe pancytopenia and splenomegaly.
Results: The interdisciplinary medical management allowed the control of the severe clinical deterioration of the patient. During the delivery, she presented postpartum hemorrhage with hypovolemic shock, which resolved with medical management. The mother and the newborn were discharged without other associated complications.
Conclusion: Interdisciplinary management is essential for handling a critically ill obstetric patient with Gaucher disease, and to achieve good maternal and perinatal outcomes.
KEY WORDS: Gaucher disease, pregnancy, thrombocytopenia, acid beta-glucocerebrosidase deficiency, lysosomal storage disorder, hepatomegaly, splenomegaly.
INTRODUCCIÓN
La enfermedad de Gaucher (EG) es la enfermedad de depósito lisosomal más común a nivel mundial, con una prevalencia que varía entre 1 a 10 por 100,000 individuos en la población general (1)(2). Es una enfermedad autosómica recesiva y consiste en una deficiencia o ausencia de la enzima beta-glucosidada ácida (GBA1), encargada de la descomposición hidrolítica de los lípidos glucocerebrosidos (glucosil-ceramida) (GC) en glucosa y ceramida. Ésta deficiencia genera una acumulación anormal de GC en el sistema monocito-macrófago (que se convierten en “células de Gaucher”) en el bazo, hígado, médula ósea, cerebro y osteoclastos (3)(4).
La EG tipo I no presenta signos neurológicos y es la forma más prevalente. La EG Tipo II se manifiesta de manera aguda con signos neurológicos severos, principalmente en la infancia, mientras la Tipo III tiene un curso crónico con deterioro progresivo (5). Durante el embarazo, la EG se asocia a una mayor incidencia de infecciones, aborto espontáneo y hemorragia postparto (HPP) (6)(7).
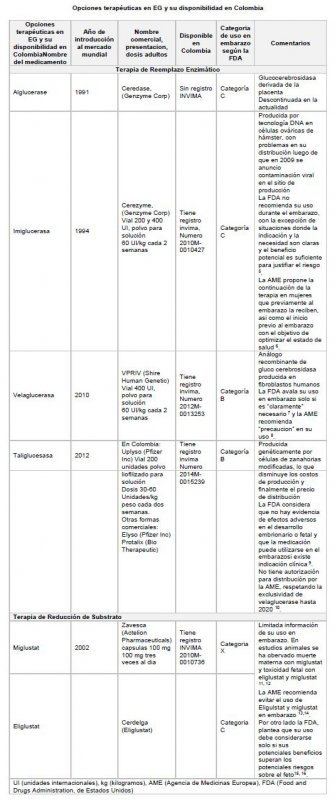
Existen dos opciones terapéuticas en la EG. La terapia de reemplazo enzimático (TRE) que consiste en suplementar la enzima activa GBA1 y la terapia de reducción de substrato (TRS) que afecta el ciclo bio-sintético de GC para disminuir en influjo hacia los lisosomas, reservada para pacientes que no pueden recibir la TRE (8)(9).
Presentamos el caso de una paciente con EG tipo I diagnosticada en el embarazo (15 semanas) quien recibió TRE con Taliglucerasa desde la semana 27, en la Unidad de Alta Complejidad Obstétrica de la Fundación Valle de Lili en Cali, Colombia
PRESENTACIÓN DEL CASO
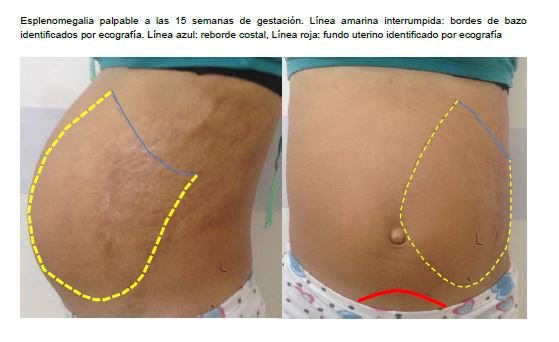
Paciente de 20 años de edad, primigesta con embarazo de 15,3 semanas, a quien de manera incidental en su control prenatal se le documenta pancitopenia con hemoglobina en 10,4 g/L, plaquetas en 42,800/μL, leucocitos 3,460/μL y esplenomegalia de 25,4 cm (figura 1).
Los estudios de autoinmunidad, pruebas hepáticas, renales e infecciosas fueron normales. La biopsia de medula ósea documentó histiocitos con sobrecarga de hierro y el cariotipo materno fue 46, XX. La actividad de la beta-glucosidasa medida por fluorometría enzimática en leucocitos (Gold estándar), resultó reducida (actividad de 0.3 nmol/h/mg, con valor de referencia: >8.7) y actividad enzimática en leucocitos de beta-glucosidasa de 0. Se hace diagnóstico de EG tipo I y se inicia tratamiento de remplazo enzimático con Taliglucerasa alfa 60 UI/kg de peso corporal, cada dos semanas, desde la semana 27 de manera ininterrumpida, con 5 dosis antes del parto.
Dentro del protocolo nacional de control prenatal se tomaron anticuerpos IgG e IgM para toxoplasma que resultaron positivos, con test de avidez de IgG baja, compatible con infección gestacional y recibió espiramicina 3.000.000 UI cada 8 horas hasta el nacimiento (19 semanas). El resultado PCR para toxoplasma en líquido amniótico fue negativo y se descartó infección fetal.
A las 37 semanas de embarazo, se realizó inducción del parto con un nivel de 49.000 plaquetas por μL. Se obtuvo un recién nacido de sexo masculino, con peso de 3271 gr, talla 47 cm y puntaje de APGAR 9-10-10. Presentó hemorragia postparto por atonía uterina con pérdida de 1100 cc de sangre cuantificada con bolsa recolectora. Recibió manejo protocolizado con masaje uterino, uterotónicos (oxitocina 100 mU/min en infusión, metilergonovina 0,2 mg intramuscular y misoprostol 800 mcg sublinguales dosis única) y ácido tranexámico (1 gr IV dosis única). Ante la persistencia del sangrado se realizó taponamiento uterino con balón hidrostático y colocación de traje anti choque no neumático con lo que se detuvo la perdida sanguínea. Requirió transfusión de una unidad de glóbulos rojos y seis unidades (1 aféresis) de plaquetas. Su evolución fue favorable y ante la solicitud de planificación familiar definitiva y la procedencia en área rural dispersa, se realizó esterilización quirúrgica por mini laparotomía en el segundo día postparto con 58.000 plaquetas por μL, egresando al cuarto día postparto. Recibió 4 dosis adicionales de Taliglucerasa en el puerperio.
No se encontraron anomalías anatómicas en el neonato y se descartó infección congénita por toxoplasmosis. El seguimiento al infante muestra desarrollo neurológico normal a los 9 meses de edad.
DISCUSIÓN
La EG es una condición infrecuente, con incidencia estimada de 1 en 100.000 individuos de la población general (10) (11). Ésta enfermedad no afecta la fertilidad, y no existe contraindicación para la gestación en ésta población, excepto por complicaciones específicas, propias de la enfermedad, como hipertensión pulmonar o alteraciones hematológicas severas.
Durante el embarazo, las principales manifestaciones clínicas son anemia, trombocitopenia, organomegalia y enfermedad ósea (12) (13). En nuestra paciente las manifestaciones hematológicas y la esplenomegalia permitieron la remisión para el manejo oportuno.
El cuidado obstétrico de pacientes con EG requiere un equipo competente. En primer lugar, el diagnóstico requiere la determinación de la actividad enzimática de la GBA1 e idealmente estudios genéticos para identificar el tipo de mutación específica en cada caso, en nuestra paciente no fue posible realizar estudio genético por perdida en el seguimiento y limitaciones de cobertura en su aseguramiento en salud. Los cambios fisiológicos del embarazo pueden aumentar la sintomatología en una paciente antes asintomática y la condición basal de la mujer con EG puede hacer más complejo el curso de un embarazo normal. Por ejemplo, la anemia y la trombocitopenia pueden agravarse en el momento del parto y en gestantes con enfermedad ósea se recomienda limitar la lactancia materna a 6 meses para disminuir el impacto en desmineralización ósea (13).
El momento de la finalización de la gestación de estas pacientes requiere consideraciones especiales. Las alteraciones hematológicas hacen que sea preferible el parto vaginal a la cesárea por el mayor riesgo de sangrado masivo y deben ser atendidas en hospitales con disponibilidad de hemocomponentes. En el caso de requerir cesárea, se debe evitar en lo posible las incisiones verticales en piel pues en el caso de hepato o esplenomegalia pueden presentarse traumatismo de estos órganos aumentados de tamaño. La visceromegalia puede también aumentar el riesgo de bronco aspiración y debe evitarse la anestesia general si el recuento plaquetario lo permite. Todas estas consideraciones fueron cuidadosamente discutidas al interior del grupo multidisciplinario institucional en ésta paciente.
Las recomendaciones actuales de las agencias de control de medicamentos americana y europea para el uso de TRE y TRS durante el embarazo se describen en la Tabla 1. La TRE está permitida durante la gestación, porque la proteína enzimática administrada no atraviesa la barrera placentaria y se inhibe a nivel gastrointestinal del recién nacido (avalando su uso en lactancia). El inicio del manejo en nuestra paciente obedeció a la trombocitopenia severa (menos de 50 mil plaquetas). Entre las opciones de manejo disponibles (tabla), Velaglucerasa y Taligluserasa pertenecen a la categoría B de riesgo de teratogenicidad (14) y por disponibilidad en nuestro medio, iniciamos Taligluserasa a la dosis reportada en la literatura (15 )
La TRS no se utiliza en embarazo porque la disminución de la producción de GC pueden afectar una variedad de funciones celulares neonatales (13) (16) (17).
En Colombia, desde el año 2010, se promulgaron medidas para garantizar la protección del Estado a la población afectada por enfermedades huérfanas (18)(19). Aunque se recomienda que la TRE se mantenga a largo plazo, en especial cuando existe organomegalia importante (20), nuestra paciente recibió solo 4 dosis adicionales Taliglucerasa en el puerperio por falta de cobertura de su asegurador y luego de 9 meses, no fue posible contactarla para seguimiento a pesar de una búsqueda activa por la red de salud regional, poniendo en evidencia algunas de las dificultades que vive la población obstetricia en los países en vías de desarrollo. Todas las instancias relacionadas con el manejo de estas pacientes debemos continuar la búsqueda de soluciones para mejorar su calidad de vida durante la maternidad.
Aunque la EG es una condición infrecuente, debe considerarse siempre entre las opciones diagnosticas en pacientes con alteración de los recuentos celulares en el hemograma y visceromegalia. Es indispensable el manejo interdisciplinario de la gestante con EG con participación activa del hematólogo, el obstetra y el genetista, así como la atención del parto en centros de alta complejidad con disponibilidad de banco de sangre y experiencia en el manejo de las posibles complicaciones relacionadas.
Agradecimientos: Los autores expresan su agradecimiento a los miembros del grupo de infectología y de hemato-oncologia por el seguimiento continuo y el apoyo científico.
Conflictos de interés: Ninguno. Las opiniones expresadas en este artículo no representan una posición institucional oficial.
BIBLIOGRAFÍA
1. Mehta A. Epidemiology and natural history of Gaucher's disease. Eur J Intern Med.
2006; 17(1) S2-S5.
2. Zimran A, Gelbart T, Westwood B, Grabowski GA, Beutler E. High frequency of the Gaucher disease mutation at nucleotide 1226 among Ashkenazi Jews. Am J Hum Genet. 1991;
49(8): 55-889.
3. Nagral A. Gaucher Disease. J Clin Exp Hepatol. 2014;
Vol. 4. No. 1. 37–50
4. Jmoudiak M, Futerman AH. Gaucher Disease: pathological mechanisms and modern management. Br J Hematol. 2005;
129:178-188.
5. Weinreb NJ, Aggio MC, Andersson HC, Andria G, Charrow J, Clarke Jt, et al. Gaucher disease type 1, Revised recommendations. Semin Hematol. 2004; 41(4):15-22.
6. Elstein Y, Eisenberg V, Granovsky-Grisaru S, Robinowitz R, Samueloff A, Zimran A, et al. Pregnancies in Gaucher diseas: A 5-year study. AM J Obstet Gynecol. 2014;
190 (2) 435-41.
7. Lau H, Belmatoug N, Deegan P, Goker-Alpan O, Schwartz I, Shankar SP, et al. Reported outcomes of 453 pregnancies in patients with Gaucher Disease: An analysis from the Gaucher outcome survey. Blood Cells Mol Dis. 2016; (16).
8. Colquicocha MM, Cucho JJ, Eyzaguirre ZR, Manassero MG, Moreno LM, Salas AK, et al. Guías para el diagnóstico y tratamiento de le Enfermedad de Gaucher.
Rev Med Hered. 2015; 26:103-121.
9. Hughes DA, Gonzalez DE, Lukina EA, Mehta A, Kabra M, Elstein D, et al.
Velaglucerase alfa (VPRIV) enzyme replacement therapy in patients with Gaucher Disease: long-term data from phase III clinical trials, Am. J. Hematol. 2015; 90 (7)
584–91.
10. DANE Proyecciones Nacionales y Departamentales de Población 2005-2020.
Bogotá, Colombia. Citado el 2 de julio de 2017. Disponible en:
https://www.dane.gov.co/files/investigaciones/poblacion/proyepobla06_20/7Proyecciones_poblacion.pdf.
11. Asociación Colombiana de Pacientes con Enfermedades de Deposito Lisosomal.
Citado el 2 de julio de 2017. Disponible en: http://www.acopel.org.co/web/.
12. Zimran A, Morris E, Mengel E, Kaplan P, Belmatoud N, Hughes DA, et a. The female Gaucher patient: the impact of enzyme replacement therapy around key reproductive events (menstruation, pregnancy and menopause), Blood Cells Mol.
2009; 43 (3) 264–288.
13. Granovsky-Grisaru S, Belmatoug N, Vom Dahl S, Mengel E, Morris E, Zimran A.
The management of pregnancy in Gaucher disease. Eur J Obstet Gynecol Reprod
Biol. 2011; 156 (1) 3-8.
14. Product Information. Elelyso (taliglucerase alfa).» Pfizer U.S. Pharmaceuticals Group, New York, NY.
15. Zimran A, Brill-Almon E, Chertkoff R, et al. Pivotal trial with plant cell-expressed recombinant glucocerebrosidase, taliglu- cerasealfa, a novel enzyme replacement therapy for Gaucher disease. Blood. 2011;118:5767-5773
16. Schiffmann R, Fitzgibbon EJ, Harris C, Deville C, Davies EH, Abel L, et al.
Randomized controlled trial of miglustat in Gaucher’s disease type 3. Ann Neurol.
2008; 64 (5): 514-522.
17.Weinreb NJ, Goldblatt J, Villalobos J, Charrow J, Cole JA, Kerstenetzky M, et al.
Long-term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J Inherit Metab Dis. 2013; 36(3);
543–553.
18. Ley 1392 de 2010. Republica de Colombia. Por medio de la cual se reconocen las enfermedades huérfanas como de especial interés y se adoptan normas tendientes a garantizar la protección social por parte del Estado colombiano a la población que padece de enfermedades huérfanas y sus cuidadores (2 de julio de 2010).
19. El Espectador. Redacción Salud. ¿Cómo esta Colombia en el tratamiento de
Enfermedades raras? Cita el 2 de julio de 2010. Disponible en:
http://www.elespectador.com/noticias/salud/como-esta-colombia-en-el-tratamiento-de-enfermedades- raras-articulo- 682225.
20. Barranger JA. Position statement: National Gaucher Foundation Medical Advisory Board, January 7. Am J Hematol. 2014 ;89:457-458. doi: 10.1002/ajh.23687