INTRODUCCIÓN
El síndrome de Li Fraumeni (SLF) es una enfermedad rara que se caracteriza por la aparición precoz de múltiples tumores malignos en un individuo que pueden debutar desde la infancia hasta la edad adulta (1). Existen dos formas clínicas: el Li Fraumeni clásico y el Li Fraumeni-like (LFL), este último con criterios menos restrictivos al momento del diagnóstico (2,3). Se hereda con carácter autosómico dominante con penetrancia incompleta, que está condicionada en el 70% de los casos por mutaciones de la línea germinal del gen TP53.
Los tumores más frecuentemente implicados en este síndrome son los sarcomas de partes blandas y osteosarcomas, cáncer de mama en mujeres premenopáusicas, tumores cerebrales, leucemia aguda y cáncer adrenocortical (4,5). Otros tumores con aumento de prevalencia entre los pacientes afectos de la enfermedad son los melanomas, tumores de células germinales, tumor de Wilms, carcinomas gástricos, colónicos, pancreáticos y de pulmón.
Presentamos una paciente con diagnóstico de SLF que acudió al Servicio de Ginecología donde fue valorada y tratada.
CASO CLINICO
Paciente de 41 años, que acude al servicio de Urgencias por cuadro de dolor y distensión abdominal en agosto de 2013. Como antecedentes familiares, su padre, que era fumador, falleció por cáncer de pulmón que fue diagnosticado a los 44 años, y su madre tuvo un cáncer de mama a los 40 años, estando actualmente sana.
Durante la anamnesis, la paciente relata que está diagnosticada de SLF, con estudio genético positivo para mutación c.587G>C p.R196 P en el gen TP53. Tiene dos hijas con estudio genético negativo para esa mutación.
En 2007 presentó una tumoración en glúteo izquierdo de unos 10 cm, la biopsia informa sarcoma de glúteo izquierdo pleomorfo (fibrohistiocitoma maligno pleomórfico), con alto índice proliferativo (Ki 67 50%). El estudio inmunohistoquímico fue: vimentina (+), CD34 (+), panqueratina AE1/AE3 (-), desmina (-), actina músculo liso (-), S100 (-), CD31 (-). Se practicó resección de glúteo izquierdo y se confirmó el diagnóstico en la pieza quirúrgica definitiva. En el Comité de Tumores se indicó tratamiento adyuvante con quimioterapia (4 ciclos de ifosfamida-adriamicina) y radioterapia sobre lecho y márgenes tumoral.
Un año más tarde en una tomografía axial computada (TAC) de control se observaron varios nódulos pulmonares sospechosos de metástasis, hallazgo que se confirmó mediante la realización de una tomografía por emisión de positrones (PET) En el Servicio de Cirugía Torácica se realizó una toracotomía derecha con resección de metástasis pulmonar; se resecaron 5 nódulos con resultado anatomopatológico de metástasis de fibrohistiocitoma maligno pleomórfico, en dos de ellos con márgenes libres. No recibió tratamiento adyuvante.
Posteriormente, en la mamografía de seguimiento de 2010, cuando la paciente tenía 38 años, se visualizó un nódulo en el cuadrante superior externo de mama derecha sospechoso, con diagnóstico en la biopsia de CDI GN2 ISB7, con estudio inmunohistoquímico de: receptores hormonales de estrógenos y progesterona 3+, herceptest 2+, con FISH que no amplificó, Ki 67 70%, p53 (+80%). Se programó cirugía conservadora de la mama con biopsia selectiva del ganglio centinela (BSGC) con resultado anatomopatológico definitivo de carcinoma ductal infiltrante bifocal de 15 y 9 mm G2 SB5 con invasión capilares y márgenes quirúrgicos libres, asociado a carcinoma intraductal de alto grado sólido con necrosis. Se localizaron dos ganglios centinelas en axila que fueron negativos para malignidad. La clasificación definitiva fue CaDI pT1c bifocal (15 y 9 mm) pN0 M0 G2 SB 8 asociado a CaID, estadío FIGO I. En Comité de Tumores de la mama, se indicó adyuvancia con quimioterapia, (ciclofosfamida + taxotere) radioterapia glandular posterior y hormonoterapia con tamoxifeno.
Un año después, en la mamografía de control, se objetivan múltiples microcalcificaciones en intercuadrantes inferiores de mama izquierda que abarcan toda la unidad glandular desde la zona retroareolar hasta plano posterior cuya biopsia asistida por vacío determinó la presencia de carcinoma intraductal de alto grado. En la ecografía axilar se informó de la existencia de una adenopatía sospechosa, por lo que se indicó estudio histológico con diagnóstico de carcinoma ductal infiltrante G2 SB6, receptores hormonales negativos, herceptest 3+, Ki 67 50%, E-cadherina (+), p53 (+80), CK 5/6 (-), CK 19 (+), por lo que se inició tratamiento neoadyuvante con paclitaxel, doxorrubicina y trastuzumab. A la finalización del mismo, se realizó mastectomía radical modificada con reconstrucción inmediata, con informe anatomopatológico de mínimo foco de CID grado intermedio retroareolar, con carcinoma in situ residual, sin evidencia de carcinoma infiltrante y ganglios axilares negativos para malignidad. La clasificación definitiva fue YpTis (R0) Yp N0 M0 (tras quimioterapia primaria) con respuesta patológica completa. Se administró radioterapia axilar de forma adyuvante, continuando con trastuzumab hasta octubre de 2012.
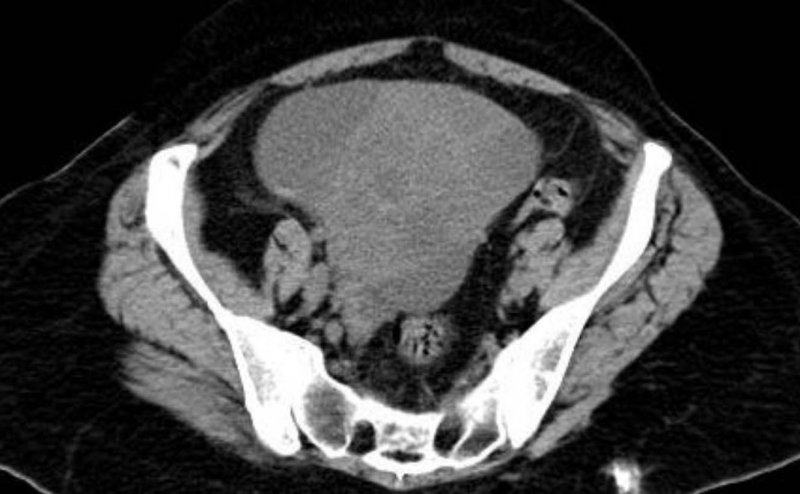
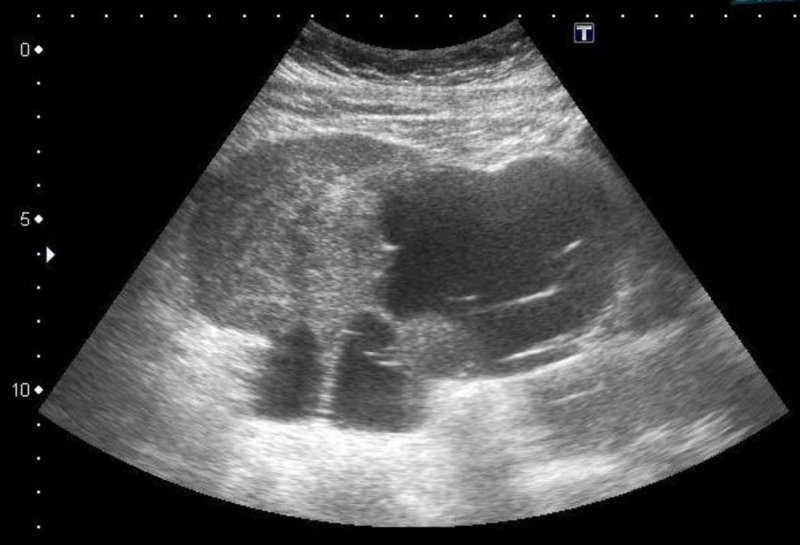
Actualmente la paciente es remitida al Servicio de Urgencias por presentar dolor abdominal que no remite con analgesia habitual, de 2 a 3 semanas de evolución y sensación de aumento de perímetro abdominal. A la exploración, se objetiva abdomen distendido, doloroso a la palpación, con signo de la oleada ascítica. En la ecografía, se visualiza una masa mixta sospechosa de 130 mm de eje máximo que parece depender de ovario derecho (Figura 1). La TAC informó de voluminosa masa central mixta quística con componente sólido 135 x 75 x 71 mm sugestiva de cáncer de ovario sin poder descartar metástasis, dados sus antecedentes (Figura 2). Los marcadores tumorales, Ca12.5, Ca19.9, Ca 15.3 y CEA eran normales. Ante la exploración, la clínica de la paciente y los hallazgos de las pruebas de imagen, se indicó cirugía, mediante laparotomía media infra y supraumbilical con hallazgos de ascitis en cantidad moderada y tumoración sospechosa de unos 10 cm heterogénea, encapsulada, que dependía de epiplón. El resto de la exploración de la cavidad abdominal y pelvis, hígado, cúpulas diafragmáticas, intestino, peritoneo, vejiga y aparato genital eran normales. No se palparon adenopatías de tamaño patológico, pélvicas ni paraaórticas. Se realizó exéresis de la masa omental que fue enviada al Servicio de Anatomía Patológica para su valoración intraoperatoria, informando de metástasis de sarcoma. Posteriormente se resecó el epiplón, con el lecho tumoral (Figura 3) y se realizó histerectomía total simple y doble anexectomía por sus antecedentes mamarios. El estudio histológico definitivo corroboró el diagnóstico emitido durante la cirugía y en el resto de piezas quirúrgicas no se halló malignidad (Figura 4).
De forma multidisciplinar, se decidió en comité de tumores, no indicar adyuvancia, y actualmente la paciente permanece libre de enfermedad bajo tratamiento con tamoxifeno iniciado en 2010 por su primer cáncer de mama.
DISCUSIÓN
El SLF se describió en 1969 a partir de datos epidemiológicos de familias con casos de sarcomas en la infancia y asociación de otros tumores (1). Se trata de un síndrome hereditario raro, clínicamente heterogéneo, con alta penetrancia sobre todo en mujeres (5), que está debido en el 70% de las ocasiones a mutaciones germinales en el gen TP53 en el cromosoma 17p13.1.
En los portadores de la mutación se estima que el riesgo de desarrollar cáncer a los 30 años es del 50% y llega al 90% a los 60 años. La probabilidad de segundos tumores es alta, especialmente de cáncer inducido por radiaciones.
El espectro de tumores que se incluyen en el SLF, que es diverso, se muestra en la Tabla I. La heterogenicidad debido a la variedad de tumores que pueden aparecer en estos pacientes hace que sea difícil plantear cuál es el seguimiento más adecuado. El cáncer de ovario no se encuadra dentro de los tumores que aparecen con mayor frecuencia dentro de este síndrome, sin embargo, aunque es raro, no es imposible la coexistencia de otros tipos de tumores en estos pacientes. Estas asociaciones condujeron a establecer unos criterios diagnósticos clínicos para el SLF y otros, menos restrictivos para el LFL (Tabla II y III).
Con respecto al cáncer de mama, un reciente estudió concluía que existe una prevalencia significativamente mayor de los tumores HER2 positivos en las pacientes con SLF en comparación con aquellas que no tienen la mutación (6). Nuestra paciente tuvo dos cánceres primitivos de mama; uno de ellos sí sobre expresó la proteína HER2.
EL TP53 es un gen supresor de tumores que codifica la proteína p53, la cual regula la apoptosis o muerte celular programada, ya que impide a la célula completar su ciclo celular si su ADN no está correctamente replicado en fase S. Por ello, las células de los pacientes con SLF podrían ser más sensibles a cualquier lesión del ADN. La p53 también está implicada en la progresión del ciclo celular después de las irradiaciones (7). Las mutaciones en Tp53 pueden modificar la capacidad de apoptosis y ser el origen de una radio resistencia tumoral (8). Este aspecto ha de tenerse en cuenta a la hora de someter al paciente a pruebas diagnósticas con radiaciones ionizantes, ya que éstas podrían desencadenar un estímulo tumoral en las células irradiadas. Por otra parte no existe contraindicación formal para administrar radioterapia o utilizar técnicas de diagnóstico que supongan radiación. El médico deberá valorar la seguridad y la necesidad de estas técnicas y sopesar el riesgo y el beneficio de las mismas.
Por todo ello, es aconsejable limitar estos procedimientos diagnósticos en los pacientes con SLF (9,10). En la paciente de nuestro caso clínico, tal vez deberíamos haber tenido más en cuenta esta circunstancia especial, ya que desde su diagnóstico ha sido sometida a numerosas pruebas de imagen que suponían un extra de irradiación: TAC, PET, mamografías, radiografías simples. Es cierto que es difícil encontrar un equilibrio entre un diagnóstico certero y precoz, sabiendo el riesgo aumentado de segundas neoplasias en los pacientes con SLF y la optimización de las pruebas diagnósticas en el seguimiento de los mismos.
Estas mutaciones pueden detectarse mediante técnicas de diagnóstico genético molecular, aunque dada la ausencia de medidas preventivas eficaces, una vez establecido el diagnóstico de la enfermedad, la utilidad del estudio genético en individuos de riesgo resulta dudosa. En la actualidad no existen programas de diagnóstico precoz que hayan demostrado eficacia a la hora de disminuir la mortalidad en los pacientes afectos. Entre las medidas de vigilancia recomendadas, sólo son efectivas aquellas que van encaminadas al seguimiento del cáncer de mama, teniendo en cuenta que la mamografía supone una irradiación que como se ha explicado puede ser nociva en estas pacientes (11).
CONCLUSIÓN
El SLF es una rara enfermedad hereditaria asociada con un riesgo incrementado de padecer ciertos tumores malignos. Su complejidad molecular, la heterogeneidad clínica y las implicaciones del consejo genético de las familias con este síndrome indican la necesidad de recomendaciones o programas protocolizados con un enfoque multidisciplinar, para poder proporcionar a estos pacientes una mejor asistencia clínica integrada.
BIBLIOGRAFÍA
1. Li FP, Fraumeni JF. Soft-tissue sarcomas, breast cancer and other neoplasms: a familial syndrome? Ann Int Med 1969;71:747-52.
2. Chompret A, Brugières L, Ronsin M, Gardes M, Dessarps-Freichey F, Abel A, et al. P53 germ line mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 2000;82:1932-7.
3. Tinat J, Bougeard G, Baert-Desurmont S, Vasseur S, Martin C, Bouvignies E, et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol 2009;27:e108-e109.
4. Li FP, Fraumeni JF Jr, Mulvihill JJ, Blattner WA, Dreyfus MG, Tucker MA, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358-62.
5. Wu CC, Shete S, Amos CI, Strong LC. Joint effects of germ-line p53 mutation and sex on cancer risk in Li-Fraumeni syndrome. Cancer Res 2006;66:8287-92.
6. Melhem-Bertrandt A, Bojadzieva J, Ready KJ, Obeid E, Liu DD, Gutierrez-Barrera AM, et al. Early onset HER2-positive breast cancer is associated with germ line TP53 mutations. Cancer 2012;118:908-13.
7. Zeng M, Narayanan L, Xu XS, Prolla TA, Liskay RM, Glazer PM. Ionozing radiation-induced apoptosis via separate PMS2 and p53 depen- dent pathways. Cancer Res 2000;60:4889-93.
8. Balmaña J, Nomdedéub J, Díezc O, Sabatéd JM, Balile A, Pericaya C, et al. Descripción de una nueva mutación germinal en el gen TP53 en una familia con síndrome de Li-Fraumeni. Asesoramiento clínico a los portadores sanos. Med Clin (Barc) 2002;119:497-9.
9. Varley JM, Evans DGR, Birch JM. Li-Fraumeni syndrome -a molecular and clinical review. Br J Can 1997;76:1-14.
10. Mirzayans R, Andrais B, Scott A, Wang YW, Murray D. Ionizing radiation-induced responses in human cells with differing TP53 status. Int J Mol Sci 2013;14:22409-35.
11. Guía de manejo del Síndrome de Li Fraumeni. Grupo de trabajo en cáncer hereditario de la Sociedad Española de Oncología Médica (SEOM). http://www.seom.org/en/publicaciones/guias-clinicas.