

2006
1
Autores:
David Mayerson B.1, Mauricio Cuello F.1, Jorge Brañes Y.1, Virginia Leiva C.2, Adriana Castiblanco G.3
Instituciones:
1Unidad de Oncología Ginecológica, Departamento de Obstetricia y Ginecología, Escuela de Medicina, Pontificia Universidad Católica de Chile. , 2Instituto de Oncología Ginecológica. , 3Departamento de Anatomía Patológica, Escuela de Medicina, Pontificia Universidad Católica de Chile.
Imágen 1:
Imágen 2:
Imágen 3:
Imágen 4:
Imágen 5:
Imágen 6:
Tipo de documento:
Casos ClínicosTumor de los cordones sexuales con túbulos anulares del ovario no asociado a síndrome de Peutz-Jeghers
Contenido del documento:
INTRODUCCIÓN
Los tumores del estroma gonadal constituyen alrededor del 7% de las neoplasias originadas en el ovario y constituyen aproximadamente el 90% de los tumores funcionantes del ovario. Dentro de este grupo de tumores, el más frecuente es el tumor de las células de la granulosa (70% de los casos). Generalmente esta variedad de tumores se presenta antes de los 40 años, en etapa temprana y habitualmente confinados al ovario. La mayoría de ellos son de bajo potencial maligno y con pronóstico favorable al largo plazo (1). En nuestra experiencia, recientemente reportada, la incidencia de tumores del estroma gonadal específico en mujer chilena fue similar a la reportada en la literatura internacional (7%) (2). De ellos, hemos ya reportado 10 casos de tumor de células de la granulosa y un caso de tumor de Sertoli Leydig asociado a virilización (3, 4). A la fecha no conocemos de reportes en la literatura nacional de casos de tumores del ovario de los cordones sexuales con túbulos anulares.
El presente trabajo tiene por finalidad comunicar un raro caso de esta entidad afectando a una mujer en edad fértil y revisar la literatura sobre el tema. El conocimiento de la historia natural de esta entidad resulta importante para el adecuado manejo de mujeres jóvenes que se puedan presentar con este tipo de tumor.
CASO CLÍNICO
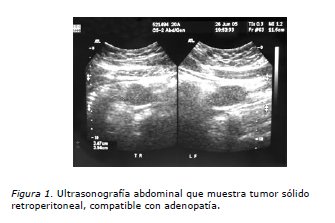
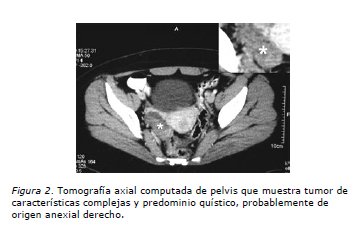
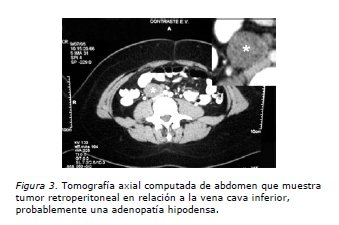
Paciente de 20 años con antecedentes de displasia esquelética parcial de extremidad superior izquierda no asociada a otras malformaciones, en control ginecológico regular hace dos años por hipermenorrea con estudio clínico y de imágenes normales. FO: 00000, sin actividad sexual. Ritmo menstrual regular 28/VI. Consulta en el Servicio de Urgencia el 26 de junio de 2005 por dolor en fosa iliaca derecha de carácter sordo, no irradiado de tres días de evolución sin otros síntomas asociados. Se realiza una ecografía abdominal que informa lesión sólida hipoecogénica de contornos bien definidos de aproximadamente 3,9 cm de diámetro mayor en situación precava infrarenal que impresiona corresponder a un ganglio aumentado de volumen sensible a la compresión con el transductor (Figura 1). No se evidencia ascitis, restos de los órganos abdominales y pelvianos normales (se consigna vejiga parcialmente distendida). Dado esos hallazgos se realiza TAC de abdomen y pelvis (9/7/2005) que informa: en relación a región anexial derecha se observa una imagen hipodensa algo heterogénea de 6,5 cm de diámetro mayor con el aspecto de corresponder a una lesión quística compleja; en situación precava inmediatamente proximal a la unión de las venas iliacas se observa masa hipodensa de 3,5 cm de diámetro mayor que podría corresponder a una adenopatía retroperitoneal. La impresión diagnóstica fue: imagen quística compleja anexial derecha (Figura 2). Masa retroperitoneal, probablemente una adenopatía hipodensa (Figura 3). Se completa el estudio con radiografía de tórax que es normal.
Es referida a ginecología oncológica, donde se realiza examen clínico limitado por obesidad de la paciente. Se solicita eco ginecológica trans rectal, hematobioquímica y marcadores tumorales.
La ultrasonografía trans rectal (18/7/05) informa: útero en AVF de forma y tamaño conservados con miometrio homogéneo de 71x33x43 mm, endometrio homogéneo de 5 mm y tipo III. Ovario izquierdo 19×11 mm de forma y aspecto normales. El ovario derecho 49x26x49 mm (32,7 cc). El aspecto es sólido con áreas hipoecogénicas, vascularizado con vasos de baja resistencia al doppler. Fondo de saco libre.
El estudio de marcadores tumorales revela: CEA 1,1 ng/ml (normal: 0 a 5 ng/ml), alfa feto <2,4 ng/ml (normal 0-12 ng/ml), CA-125 16,2 U/ml (normal: <35 U/ml), Beta HCG <0,1 (normal: <10). Se solicito además en el estudio diferencial serología para yersinia sp (3 variantes) negativo, virus Ebstein Barr (IgG positivo, IgM negativo).
Hemograma: hematocrito 43,5%, leucocitos 5.110 y sin desviación a izquierda, plaquetas: 238.000, VHS: 3 mm/hora, PCR normal, LDH normal.
Dado los hallazgos de imágenes se decide laparotomía exploradora que se realiza el 4/8/2005.
Se realiza laparotomía supra e infraumbilical. Citología por lavado (ausencia de ascitis). Los hallazgos intraoperatorios revelan: ovario derecho tumoral, sólido, libre, 7×5 cm, y se procede a anexectomía derecha. Biopsia contemporánea: tumor del estroma gonadal (diagnóstico diferencial: entre tumor de la granulosa y tumor de cordones sexuales túbulo anulares). Se procede a identificar nódulo descrito en ECO y TAC de abdomen que corresponde a un nódulo de la raíz del mesenterio del intestino delgado lográndose su extirpación completa. Se envía nódulo a biopsia rápida que demuestra histología equivalente al tumor primario. Se procede luego a linfadenectomía pelviana ipsilateral, omentectomía infracólica. Resto de la exploración abdominal y retroperitoneal sin evidencias de enfermedad. Evolución postoperatoria precoz y tardía: normal, sin incidentes
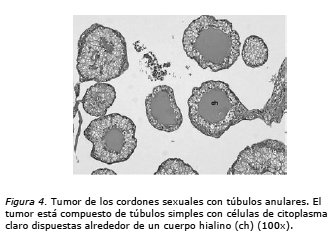
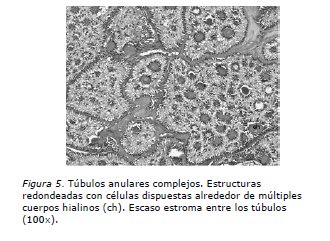
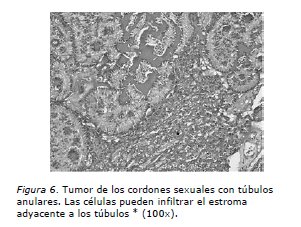
El estudio de anatomía patológica informa: Tumor de los cordones sexuales con túbulos anulares del ovario derecho. Nódulo mesentérico constituido por tumor ya descrito. Ganglios pelvianos sin compromiso tumoral y citología peritoneal negativa (Figuras 4, 5 y 6).
Discutido el caso, se etapifica como tumor del estroma gonadal específico en etapa avanzada (etapa IIIc). Presentado el caso en comité oncológico se decide seguimiento clínico y de imágenes.
DISCUSIÓN
El tumor de los cordones sexuales con túbulos anulares del ovario (TCSTA) constituye una entidad rara dentro de los tumores del estroma gonadal específico. Algunas series han reportado una incidencia de 6% de todos los tumores del estroma gonadal, lo que equivale al 0,4% de los tumores malignos del ovario (1). La primera descripción de esta entidad fue hecha por Scully en 1970 (5). Lo distintivo en este tumor era la presencia de elementos celulares intermedios entre tumor de células de la granulosa y de células de Sertoli. Dentro de esta entidad se distinguen dos subtipos. Los TCSTA asociados al síndrome de Peutz-Jeghers (SPJ) y aquellos no asociados a dicho síndrome (1, 6). El SPJ es una enfermedad genética autosómica dominante asociada al desarrollo de pólipos gastrointestinales con un riesgo elevado de desarrollar cáncer digestivo (colon, estómago, páncreas, etc.), ginecológico (ovario, cuello uterino y mama) (7). Dicho mayor riesgo se debería a la pérdida de un gen supresor conocido como STK11 (una kinasa del tipo serina-treonina) localizado en el brazo corto del cromosoma 19 (8). Aproximadamente 1 de cada 3 de los TCSTA se asociaría a este síndrome (9). Característicamente los TCSTA asociados a SPJ son tumores pequeños (la mayoría microscópicos), multifocales, calcificados, bilaterales, de presentación en la cuarta década de la vida. Por contraste los TCSTA no asociados al síndrome son más grandes, unilaterales, rara vez calcificados o multifocales y de presentación en la tercera década de la vida (6, 10). En nuestra paciente el tumor se presentó al inicio de la tercera década de la vida, siendo unilateral, de diámetro mayor ~5 cm, unifocal y sin áreas calcificadas. Rara vez los TCSTA se presentan dentro de la primera década de la vida. Sin embargo, en los casos reportados se describe la ocurrencia de pubertad precoz isosexual (9).
La forma de presentación es habitualmente por alteración de flujos rojos con irregularidad menstrual y episodios de metrorragia. En TCSTA sin SPJ, la menometrorragia seguida de períodos de amenorrea es bastante característica. A ello se agrega, como forma de presentación, la pubertad precoz y la metrorragia postmenopáusica cuando la entidad se presenta en los extremos de la vida. Todas manifestaciones propias de un tumor hormonalmente funcionante. No infrecuentemente se puede presentar, producto del desorden hormonal, habitualmente hiperestrogénico, hiperplasia endometrial o pólipos endometriales (estos últimos más frecuentes en los TCSTA asociados a SPJ). Dichas alteraciones se corrigen una vez extirpado el tumor. Menos frecuentemente el TCSTA se presenta con dolor abdominal o sensación de plenitud. En esos casos, el dolor obedece al tamaño del tumor anexial o patología pelviana asociada. En nuestra paciente la forma de presentación fue el dolor en la fosa iliaca derecha, sitio donde se ubicaba el tumor anexial. En los TCSTA asociados a SPJ, una forma de presentación puede ser la intusucepción del intestino grueso por la presencia de pólipos colonicos, frecuentemente presente en las pacientes con SPJ (1).
Típicamente el TCSTA asociado a SPJ es hallazgo en la anatomía patológica dado que se trata de tumores microscópicos. En contraste los TCSTA no asociados al síndrome son palpables al examen abdominal o vaginal o detectados mediante exámenes radiológicos. En este caso el tumor fue hallazgo de la ultrasonografía y no era palpable dada la obesidad de la paciente.
Del punto de vista patológico, los TCSTA asociados a SPJ son tumores sólidos de color amarillo. Por contraste, los TCSTA no asociados al síndrome son tumores sólido-quísticos o de predominio quístico. Típicamente presentan a la microscopia túbulos anulares simples y complejos (Figuras 4 y 5). En nuestro caso, el tumor al corte era una formación quística unilocular ocupada por tejido tumoral amarillo ocre. A la microscopia se identificaba el aspecto clásico de túbulos anulares simples y complejos con menor proporción de áreas sólidas, papilas y algunos quistes simples. El índice mitótico era de 0 a 4 mitosis por cada 10 campos de aumento mayor. El tumor no comprometía la cápsula ovárica ni presentaba permeaciones vasculares. El nódulo mesentérico presentaba las mismas características del tumor primario y no se reconocía en él la presencia de tejido ganglionar.
Así, considerando las características clínicas (forma de presentación, edad de la paciente), del tumor a la macroscopa y microscopia parece más probable que se trate de un TCSTA no asociado a SPJ. No existiendo antecedente familiar del síndrome se hace menos probable la asociación y más raro que se trate de una forma esporádica del síndrome.
Los TCSTA asociados a SPJ son de curso benigno. Sin embargo, la asociación del SPJ con adenoma maligno de cuello, en un 15% de los casos, les confiere un mal pronóstico. Dicha entidad corresponde a un cáncer de cuello uterino de presentación a edad temprana, agresivo y de mal pronóstico. Aproximadamente 20% de los TCSTA no asociados al síndrome tendrá un curso maligno (con enfermedad extendida fuera del ovario y recurrencia de la enfermedad). Dicho riesgo se correlacionaría con el tamaño y el índice mitótico del tumor primario (9).
Respecto del tratamiento de esta entidad, tanto los TCSTA con o sin PJS son tratados primariamente con cirugía. Indispensable resulta la remoción de los ganglios de la cadena pelviana ipsilateral y periaórtica dado que es la forma de diseminación más frecuentemente reportada. En los casos de TCSTA sin PJS, no se requeriría de biopsia en cuña del ovario contralateral si este es normal. Por contraste los TCSTA con PJS requieren de la anexectomía bilateral considerando la bilateralidad y multifocalidad asociadas (1). No existe información sobre la utilidad de la omentectomía, biopsias peritoneales o citología peritoneal más si se considera lo poco frecuentes que son los casos de TCSTA con enfermedad diseminada intraabdominalmente.
Existe escasa información sobre terapias complementarias en casos de enfermedad avanzada particularmente si se ha alcanzado la remoción completa de la enfermedad (incluido compromiso ganglionar). La mayoría de las series reportan casos con enfermedad confinada al ovario. El curso lento y las recurrencias tardías (varios años posterior al tratamiento primario), han hecho planteable la opción de seguimiento y tratamiento al momento de la recurrencia. En los casos de recurrencia las alternativas han incluido la cirugía de resección de la enfermedad (incluidas citoreducciones repetidas), la radioterapia dirigida al sitio con enfermedad, y la quimioterapia. Con todas las modalidades ha habido respuesta y se ha reportado sobrevida de años (1, 9, 11). En nuestra paciente, dado que se alcanzó remoción completa de la enfermedad se decidió seguimiento clínico estricto. Puls y cols (12), reportaron un caso de enfermedad recurrente intrabdominal con un gran tumor que fue manejado primariamente con cirugía, a 9 meses de la cirugía presentó una segunda recurrencia con metástasis hepática; dicha recurrencia fue tratada con quimioterapia (una combinación de etopósido, bleomicina y cisplatino) alcanzando respuesta completa (confirmada por re-laparotomía). Así, pareciera razonable reservar terapias complementarias al momento de la recurrencia. Por tanto, decidimos reservar estas opciones ante una eventual recurrencia.
En cuanto al seguimiento, éste se ha limitado al examen clínico periódico y estudio de imágenes. Recientemente, algunos reportes sugerirían la utilidad de dos marcadores hormonales, el factor inhibidor mülleriano e inhibina. Existiría correlación entre los niveles de dichos marcadores tanto con el status clínico de la paciente como con la respuesta a terapias de rescate (quimioterapia) (12). Basado en ello, hemos recomendado a nuestra paciente el realizar inhibina en forma periódica como parte de su seguimiento.
Por último, creemos importante comentar la necesidad de explorar la historia familiar en búsqueda de neoplasias, particularmente digestivas, a fin de descartar la existencia de un SPJ. Así también, cabe señalar que frente a casos de histología inhabitual es necesario interconsultar patólogos con mayor experiencia en el tema quienes puedan confirmar el diagnóstico. Cabe destacar sobre este punto que existen casos reportados en la literatura de sarcomas de tubo digestivo (originados en el estómago) los cuales pueden remedar las características de un tumor de células de Sertoli. Por ello, junto con revisar la patología es necesario recomendar la evaluación del tubo digestivo (eventualmente realizar endoscopia).
CONCLUSIONES
Los TCSTA no asociados a SPJ son tumores raros, que afectan a mujeres en edad fértil y que son suceptibles de un manejo conservador dado que al momento de su presentación habitualmente están confinados a un ovario. El tratamiento primario es la cirugía y debe incluir la anexectomía unilateral y linfadenectomía pelviana ipsilateral y periaórtica más exploración cuidadosa del abdomen y ovario contralateral. El desbalance hormonal asociado justifica la evaluación intraoperatoria del endometrio a fin de descartar una hiperplasia endometrial (biopsia de endometrio). El curso lento y las recurrencias tardías hacen planteable el seguimiento clínico y reservar las terapias complementarias al momento del rescate. En el seguimiento clínico puede ser útil la medición de inhibina y factor inhibidor mülleriano.
BIBLIOGRAFÍA
1. Gershenson DM, Hartmann LC, Young RH. Ovarian sex cord-stromal tumors. In: Hoskins WJ, Perez C, Young RH, Barakat R, Markman M, Randall M, eds. Principles and practice of gynecologic oncology. Volume 1. Fourth Edition ed. Philadelphia: Lippincott Williams and Wilkins, 2005; 1011-34. [ Links ]
2. Cuello M, Merino P, Etchegaray A, Ortega J, Pomés C, Barrena N, Gejman R. Distribución de la patología anexial en mujer chilena: experiencia de la Universidad Católica de Chile. Rev Chil Obstet Ginecol 2004; 69: 429-40. [ Links ]
3. Cuello M, Merino P, Etchegaray A, Pomés C, Gejman R, Pires Y, Brañes J. Tumor de células de la granulosa. Rev Chil Obstet Ginecol 2003; 68: 376-86. [ Links ]
4. Correa R, Cuello M, Gejman R, Merino P, Etchegaray A, Carrasco M, Brañes J. Tumor de células de Sertoli y Leydig del ovario. Rev Chil Obstet Ginecol 2003; 68: 399-406. [ Links ] 5. Scully RE. Sex cord tumor with annular tubules a distinctive ovarian tumor of the Peutz-Jeghers syndrome. Cancer 1970; 25: 1107-21. [ Links ]
6. Young RH, Scully RE: Sex cord-stromal, steroid cell, and other ovarian tumors with endocrine, paraendocrine, and paraneoplastic manifestations. In: Kurman RJ, ed. Blaustein’s pathology of the female genital tract. Baltimore: Springer-Verlag, 2002; 905-966. [ Links ]
7. Abdel-Rahman WM, Peltomaki P. Molecular basis and diagnostics of hereditary colorectal cancers. Ann Med 2004; 36: 379-88. [ Links ]
8. Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 1998; 18: 38-43. [ Links ]
9. Young RH, Welch WR, Dickersin GR, Scully RE. Ovarian sex cord tumor with annular tubules: review of 74 cases including 27 with Peutz-Jeghers syndrome and four with adenoma malignum of the cervix. Cancer 1982; 50: 1384-02. [ Links ]
10. Popiolek D, Demopoulos R. Gonadal sex cord-stromal tumors. In: Altchek A, Deligdisch L, Kase N, eds. Diagnosis and management of ovarian disorders. Volume 1. San Diego: Elsevier Science, 2003; 111-26. [ Links ]
11. Shen K, Wu PC, Lang JH, Huang RL, Tang MT, Lian LJ. Ovarian sex cord tumor with annular tubules: a report of six cases. Gynecol Oncol 1993; 48: 180-4. [ Links ]
12. Puls LE, Hamous J, Morrow MS, Schneyer A, MacLaughlin DT, Castracane VD. Recurrent ovarian sex cord tumor with annular tubules: tumor marker and chemotherapy experience. Gynecol Oncol 1994; 54: 396-401. [ Links ]


