2018
1
Autores:
Taqua Blanca R. 1, Lorena Sabonet M. 2, Patricia Perez-Moneo P. 1, Maria Luisa Perez E. 3, Amparo Sanchís. 4, Maria Reyes Balanzá C. 5
Imágen 1:
Imágen 2:
Imágen 3:
Tipo de documento:
Casos ClínicosDIAGNOSTICO PRENATAL EN SINDROME DE CORNELIA DE LANGE A PROPÓSITO DE 2 CASOS.
Contenido del documento:
RESUMEN
El Síndrome de Cornelia de Lange (SCdL) es un trastorno hereditario del desarrollo con transmisión dominante, aunque la mayoría de los casos son esporádicos. La prevalencia es variable oscilando entre 1/10.000-1/100.000 nacimientos. Se caracteriza por un fenotipo facial distintivo, anomalías en las extremidades superiores y retraso del crecimiento y psicomotor.
El diagnóstico prenatal de este síndrome está limitado a la detección de anomalías mayores, ya que los rasgos fenotípicos distintivos del mismo no son fácilmente detectables. Suele cursar con aumento de la translucencia nucal, higroma quístico y valores de PAPP-A bajos en el primer trimestre de la gestación; retraso del crecimiento intrauterino, retromicrognatia, anomalías con grado variable de severidad de las extremidades superiores y otras anomalías cardiovasculares, gastrointestinales o genitourinarias que condicionan el pronóstico fetal.
Se presentan los hallazgos ecográficos de dos casos con sospecha de afectación por el SCdL, y la correlación entre los mismos y los hallazgos en la necropsia. Al establecer la sospecha diagnóstica de forma retrospectiva, en los casos presentados no fue posible estudiar la presencia de mutaciones genéticas asociadas con el SCdL.
A pesar de los avances en el diagnóstico genético de este síndrome, la base genética del mismo es todavía desconocida en alrededor del 30% de los pacientes, lo que sugiere la contribución de otros genes y/o factores ambientales en su etiología.
PALABRAS CLAVE: Cornelia de Lange, Diagnóstico prenatal, necropsia fetal.
SUMMARY
Cornelia de Lange Syndrome (CdLS) is an hereditary developmental disorder with dominant condition, although most cases are sporadic. The prevalence is variable ranging from 1/10,000 to 1/100,000 live births. It is characterized by a distinct facial phenotype, upper limb abnormalities, growth retardation and severe mental retardation.
Prenatal diagnosis of this syndrome is limited to detecting major abnormalities, since characteristic facial features aren’t easily detectable. Is usually associated with increased nuchal translucency, cystic hygroma and low PAPP-A levels in first trimester of pregnancy; intrauterine growth retardation, retromicrognathia, anomalies with varying degrees of severity of upper limbs and other cardiovascular, gastrointestinal or genitourinary abnormalities that affect fetal prognosis.
The sonographic findings of two cases with suspected involvement by CdLS, and the correlation between them and necropsy findings are presented. Since the suspected diagnosis was established retrospectively in the presented cases, it wasn’t possible to study the association with CdLS gene mutations.
Despite advances in genetic diagnosis of this syndrome, the genetic basis of it still unknown in about 30% of patients, suggesting the contribution of other genes and/or environmental factors in its etiology.
KEY WORDS: Cornelia de Lange, prenatal diagnosis, fetal necropsy.
El Síndrome de Cornelia de Lange (SCdL) es un trastorno hereditario del desarrollo con transmisión dominante, y con menor frecuencia ligada al X, aunque la mayoría de los casos son esporádicos. La prevalencia es variable oscilando entre 1/10.000-1/100.000 nacimientos (1, 2, 3). En la actualidad se conocen varios genes causales que codifican proteínas reguladoras o estructurales del complejo de cohesinas, implicados en el desarrollo del SCdL. La mutación en el gen NIPBL está presente en cerca del 60% de los pacientes afectos de SCdL (3). Mutaciones en SMC1A, SMC3, RAD21 y HDAC8 (las dos últimas ligadas al cromosoma X) han sido también detectadas en una pequeña proporción de pacientes afectos de este síndrome (6, 7, 8).
Se caracteriza por un fenotipo facial distintivo, anomalías en las extremidades superiores y retraso del crecimiento y psicomotor. El fenotipo más grave sólo se ha hallado en pacientes con mutaciones en el gen NIPBL.
El diagnóstico prenatal de este síndrome está limitado a la detección de anomalías mayores, ya que los rasgos fenotípicos distintivos del mismo no son fácilmente detectables. Suele cursar con retraso del crecimiento intrauterino, retromicrognatia, anomalías con grado variable de severidad de las extremidades superiores y otras anomalías cardiovasculares, gastrointestinales o genitourinarias que condicionan el pronóstico fetal (3,4).
MATERIAL Y MÉTODO
Se presentan los hallazgos ecográficos de dos casos con sospecha de afectación por el SCdL, y la correlación entre los mismos y los hallazgos dismórficos postmortem y necrópsicos.
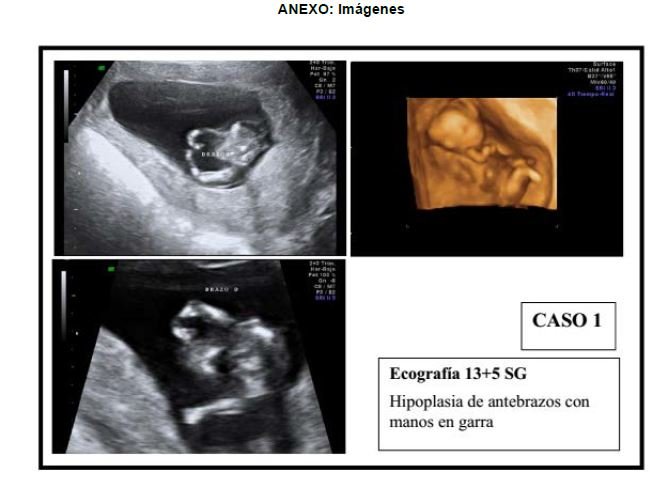
El primer caso se trata de una mujer de 33 años de edad sin antecedentes de interés ni hábitos tóxicos, primigesta, gestación espontánea. En la ecografía de primer trimestre destaca una TN de 4.5mm (>p99) y una sospecha de hipoplasia de antebrazos y manos, así como un corazón con sospecha de ventrículo único, que se confirma en las siguientes exploraciones ecográficas.
Se realiza una biopsia corial que informa de un cariotipo alterado, 47,XY+15, recomendando comprobación posterior mediante amniocentesis, que informa de un cariotipo normal, 46,XY, con trisomía 15 confinada a la placenta. Dados los hallazgos ecográficos, la pareja opta por la interrupción legal de la gestación a las 16 semanas.
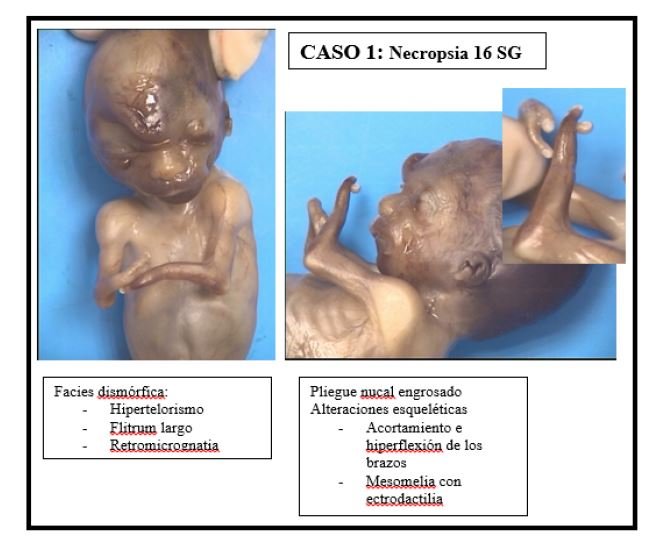
Los hallazgos de la necropsia confirman los ecográficos. Se aprecia una hipoplasia de miembros superiores, con mesomelia y ectrodactilia tipo II, un corazón de ventrículo único. Los rasgos fenotípicos faciales son característicos, con cejas en arco, hipoplasia nasal, micrognatia e hipertelorismo.
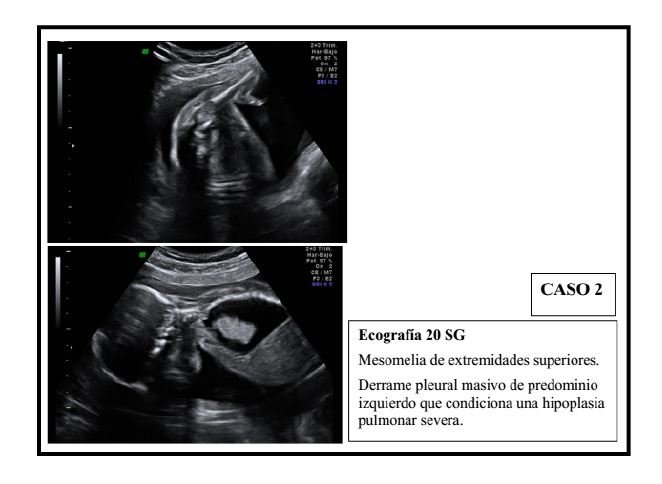
El segundo caso es una mujer de 34 años de edad, sin antecedentes médicos ni hábitos tóxicos. Secundigesta, con un hijo previo sano. Es derivada a nuestro centro a las 20 semanas de gestación ante el hallazgo, en la ecografía morfológica fetal, de mesomelia en extremidades superiores y derrame pleural masivo de predominio izquierdo que condiciona una hipoplasia pulmonar severa, y posiblemente también cardíaca. No se observan otras anomalías anatómicas.
El cariotipo fetal obtenido mediante amniocentesis informa de un feto de sexo masculino cromosómicamente normal (46,XY). Como en el caso anterior, la pareja opta por la interrupción legal de la gestación a las 22 semanas.
En la necropsia se aprecia una mesomelia de extremidades superiores, un derrame pleural bilateral de predominio izquierdo y una hipoplasia pulmonar y cardíaca, siendo éste último anatómicamente normal. Así mismo, presenta rasgos dismórficos faciales, destacando orejas de implantación baja, filtrum largo y retromicrognatia.
RESULTADOS
En los casos presentados, la evaluación dismorfológica y el estudio anatomopatológico fetal fue clave en el diagnóstico, ya que las limitaciones del estudio morfológico ecográfico y genético (cromosómico) no orientaron al diagnóstico de dicha entidad. Fueron principalmente los rasgos fenotípicos faciales y la alteración en las extremidades superiores lo que permitió establecer la sospecha diagnóstica.
Al establecer la sospecha diagnóstica de forma retrospectiva, en los casos presentados no fue posible estudiar la presencia de mutaciones genéticas asociadas con el SCdL.
DISCUSIÓN
El diagnóstico prenatal está sufriendo un cambio continuo en los últimos años, siendo posible el diagnóstico de anomalías fetales a edades gestacionales cada vez más tempranas. Sin embargo, el diagnóstico está limitado a las anomalías con manifestaciones morfológicas. El SCdL es una entidad poco frecuente con un grado variable de expresividad; no obstante, cuando presenta malformaciones mayores es posible el diagnóstico prenatal.
La confirmación diagnóstica es clásicamente postnatal, ya que los rasgos clínicos más característicos del síndrome son faciales y difícilmente detectables por ecografía. No obstante, cuando asocia alteración en los miembros superiores, debe plantearse el diagnóstico diferencial con este síndrome.
Actualmente el diagnóstico prenatal de este síndrome es factible, aunque no deja de ser un reto diagnóstico. Cuando se establece la sospecha clínica de forma prenatal, es posible realizar el estudio genético de material fetal obtenido mediante Biopsia Corial (a las 10-12 semanas de gestación), o mediante Amniocentesis (a las 16-18 semanas de gestación). La sospecha diagnóstica se establece mediante el estudio ecográfico habitual en gestaciones de bajo riesgo (triple screening a las 11-13+6 semanas y ecografía morfológica fetal a las 20-22 semanas). Los hallazgos analíticos y ecográficos reportados en la literatura son los siguientes (1, 4):
– Aumento de la translucencia nucal, higroma cístico y valores de PAPP-A bajos en el primer trimestre de la gestación (9, 10).
– Retraso de crecimiento, de inicio típicamente en el segundo trimestre.
– Rasgos faciales característicos: micrognatia, labio superior prominente, puente nasal deprimido con narinas antevertidas, filtrum largo, hipertelorismo.
– Alteración en los miembros superiores; se presentan en aproximadamente un tercio de los pacientes afectos de este síndrome, y presentan un grado de afectación variable que oscila desde la oligodactilia, hasta la ausencia de cúbito o de antebrazo, con dedos directamente sobre el codo (11).
El estudio anatomopatológico y la valoración dismorfológica de los fetos abortados es esencial para llegar a establecer una sospecha diagnóstica que permita realizar un correcto asesoramiento reproductivo a las parejas. En los casos presentados, los hallazgos ecográficos y la normalidad del estudio genético no fueron suficientes para establecer la sospecha diagnóstica de SCdL.
El hecho de que se trate de una condición no letal y que asocia múltiples anomalías y retraso del crecimiento y psicomotor, hace que el diagnóstico prenatal tenga un impacto todavía mayor en el asesoramiento de las parejas.
En los casos diagnosticados clínicamente, la tasa de detección de la mutación del gen NIPBL y SMC1L es del 50-60% y del 4%, respectivamente (6,12). En presencia de alteraciones ecográficas sugestivas estaría indicada la realización del test genético. El hecho de no encontrar anomalías genéticas no descarta el diagnóstico de esta entidad.
Aunque la mayoría de las mutaciones son de novo, y el diagnóstico genético no altera la decisión de continuar con la gestación en los progenitores, la confirmación genética es importante por varios motivos. En primer lugar, porque en presencia de retromicrognatia y anomalías severas de las extremidades superiores, el rendimiento del diagnóstico genético es muy alto, siendo cercano al 60% en algunas series (3); y en segundo lugar, porque se ha descrito mosaicismo germinal en progenitores clínicamente no afectados de fetos con variantes patogénicas en NIPBL que condicionan un riesgo de recurrencia del 1.5% en su descendencia.
Sin embargo, el diagnóstico de este síndrome sigue siendo eminentemente clínico debido a varios motivos: el coste del estudio genético, la heterogeneidad genética (deben buscarse mutaciones en los cinco genes descritos (SMC1A, SMC3, RAD21 y HDAC8) o la naturaleza esporádica de las mismas, entre otros.
A pesar de los avances en el diagnóstico genético de este síndrome, la base genética del mismo es todavía desconocida en alrededor del 30% de los pacientes, lo que sugiere la contribución de otros genes y/o factores ambientales en su etiología (5).
BIBLIOGRAFÍA
1. Karen Chong, Sarah Keating, Stephanie Hurst, Anne Summers, Howard Berger, Gareth Seaward, et al. Cornelia de Lange Syndrome (CdLS): prenatal and autopsy findings. Prenat Diag, 2009;29: 489-494
2. Dinah M. Clark, Ilana Sherer, Matthew A Deardorff, Janice L.B. Byrne, Kathleen M. Loomes, Malgorzata J.M. Nowaczyk, et al. Identification of a Prenatal Profile of Cornelia de Lange Syndrome (CdLS): a review of 53 CdLS pregnancies. Am J Med Genet Part A, 2009;158:1848-1856
3. M. A. Dempsey, A. E. Knight Johnson, B. S. Swope, J. S. Moldenhauer, H. Sroka, k. Chong. Molecular confirmation of nine cases of Cornelia de Lange syndrome diagnosed prenatally. Prenat Diag, 2014;34:163-167
4. Dave Usha, Shetty Dhanlaxmi. Mutational Screening and Prenatal Diagnosis in Cornelia de Lange síndrome. J Obstet Gynecol India 2014;64:27-31
5. Boyle M.I., Jespersgaard C., Brøndum-Nielsen K., Bisgaard A.-M. Tümer Z. Cornelia de Lange síndrome. Clinic Genet, 2015;88:1-12
6. Musio A, Selicorni A, Focarelli ML, et al. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet, 2006;38:528-530.
7. Matthew A. Deardorff, Jonathan J. Wilde, Melanie Albrecht, Emma Dickinson, Stephanie Tennstedt, Diana Braunholz, et al. RAD-21 mutations cause a human cohesinopaty. Am J Hum Genet, 2012;90(6):1014-1027
8. Matthew A. Deardorff, Masashige Bando, Ryuichiro Nakato, Erwan Watrin, Takehiko Itoh, Masashi Minamino, et al. HDAC8 mutations in cornelia de Lange síndrome affect the cohesin acetylation cycle. Nature, 2012;489(7415):313-317
9. Arbuzova S, Nikolenko M, Krantz D, et al. Low first trimester pregnancy-associated plasma protein-A and Cornelia de Lange Syndrome. Prenat Diagn, 2003;23:864.
10. Huang WH, Porto M. Abnormal first trimester fetal nuchal translucency and Cornelia de Lange syndrome. Obstet Gynecol, 2002;99:956-946.
11. Kline AD, Krantz ID, Sommer A, el al. Cornelia de lange syndrome: clinical review, diagnostic and scoring systems and anticipatory guidance. Am J Med Genet, 2007;143ª:1287-1296.
12. Bork G, Redon R, Sanlaville D, et al. NIPBL mutations and genetic heterogeneity in Cornelia de Lange syndrome. J Med Genet, 2004;41:1-6.


