2018
2
Autores:
René Rivera Z.1, Jorge Delgado D.1, Vicente Painel P.1, Raúl Barrero P.1, Angélica Larraín H.a
Instituciones:
(1) Servicio de Ginecología y Obstetricia, Hospital Clínico Félix Bulnes.
(a) Matrona, Facultad de Ciencias Médicas, Escuela de Obstetricia, Universidad de Santiago de Chile.
Imágen 1:
Imágen 2:
Imágen 3:
Imágen 4:
Imágen 5:
Tipo de documento:
DocumentosMecanismo de infección y transformación neoplásica producido por virus papiloma humano en el epitelio cervical
Contenido del documento:
El virus papiloma humano (VPH) corresponde a un virus ADN, que infecta piel y mucosas, altamente especie específico (1). Actualmente la infección por VPH es reconocida como la enfermedad de transmisión sexual más frecuente en el mundo (2). Produce una amplia variedad de lesiones, de especial importancia neoplasias intraepiteliales del tracto genital inferior y cáncer (3).
Más de cien tipos de VPH han sido identificados, actualmente alrededor de 33 de ellos han sido vinculados a lesiones genitales intraepiteliales e invasoras (4). De particular importancia son aquellos clasificados como de alto riesgo oncogénico (15 tipos), capaces de llevar al desarrollo de cáncer (5).
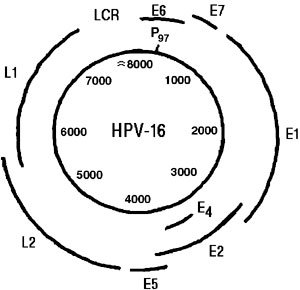
Si se toma como modelo el VPH 16, es posible esquematizar el mapa genómico (Figura 1). Presenta una estructura de aproximadamente 8.000 pares de bases, destacando una región temprana (E), una región tardía (L), una región de control largo (LCR) y 8 regiones de lectura abierta (ORF) (6).
En trabajos que intentan explicar el modelo de infección por VPH de alto riesgo, utilizando partículas tipo virus, se ha evidenciado que durante la actividad sexual, el microtrauma del epitelio genital, en particular en la zona de transformación del epitelio cervical, permite la exposición de las células basales en activa proliferación, a los diferentes tipos de HPV, permitiendo la unión entre el receptor de la célula basal con la proteína de la cápside viral L1, a nivel de su extremo carboxi terminal (7). Dicho receptor ha sido asociado estructuralmente con Heparán Sulfato para los tipos virales 16 – 33 y con Alfa-6-Integrina para VPH 6 (8,9). Una vez unido el virus a la superficie celular, se produce su internalización al citoplasma de la célula huésped, proceso que ha sido identificado como endocitosis (9). Dos sistemas han sido reconocidos; el primero involucra un complejo proteico llamado Clatrina (10), utilizado por los tipos 16 y 18; el segundo, utiliza un grupo de proteínas principalmente Caveolina, denominado endocitosis por caveolas, en el que participa el VPH 31 (11). Posterior a la endocitosis, existe evidencia en modelos de infección por partículas virales tipo 11 y 16 que la cápside viral de 55 nm de diámetro experimenta degradación en el citoplasma celular, a través de un proceso de reducción química que daña los puentes disulfido que estabilizan la cápside, originando capsómeros y monómeros, los cuales son transportados al núcleo junto a pequeños fragmentos del ADN viral, pudiendo atravesar los poros nucleares de un diámetro aproximado a 39 nm, con ello el genoma viral y las proteínas de la cápside participarían en los procesos de transcripción génica, replicación del ADN y maduración de viriones (12,13).
Es posible definir una población viral no productiva, localizado en el estrato basal, en la cual se mantiene la replicación del ADN viral en un número de copias bajo (30-50 copias por célula infectada), en forma extracromosómica, llamados episomas que se estructuran en base a histonas y material genético (14,15,16). Se postula que durante esta etapa se aseguraría que el ADN viral se distribuya difusamente por las células basales proliferantes y que al mantener un número reducido de copias se impediría la activación de la respuesta inmune (17). Las células basales proliferantes migran a los estratos parabasal y espinoso, amplificándose la expresión de genes virales tempranos a través de la región no codificante (URR), los cuales permiten producir ADN a cientos de copias por célula (18); ésta etapa en el ciclo viral es conocida como la fase vegetante, proliferante o productiva (19). En este proceso de replicación del ADN viral participan un grupo de proteínas, cuyas características principales se mencionan a continuación.
E1 corresponde a una proteína multimérica con actividad ATPasa y helicasa, capaz de recocer sitios específicos en el ADN viral, llamadas regiones de origen de replicación (ori), formando un complejo de iniciación con la proteína E2, ciclinas (principalmente E), kinasas dependiente de ciclinas y ADN polimerasa a, en la región p68, participando además proteínas de la célula huésped (20,21).
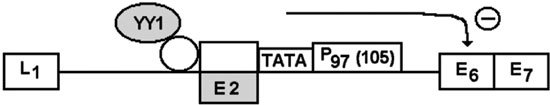
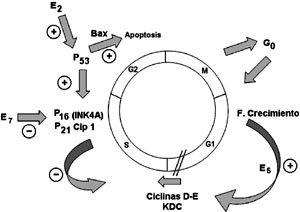
E2 corresponde a una proteína dimérica que forma un complejo proteico con E1 antes mencionado. En este proceso, E2 es fosforilado por kinasas en los residuos aminoacídicos de serina 298 y 301, regulando su unión con E1 (22). Además, E2 reprime al promotor P97 (VPH 16) o P105 (VPH 18), ubicado próximo al gen TATA box, encargado de la transcripción de las proteínas E6 y E7, reduciendo de esta manera la síntesis de estas proteínas (23) (Figura 2). También se ha demostrado que E2 activa directamente la síntesis de la proteína P53 lo que produciría finalmente la detención del ciclo celular en G1 y apoptosis (24).
La proteína E1-E4 es la mayoritariamente expresada en epitelios infectados por VPH (aproximadamente el 30% del contenido proteico celular), se sintetiza como proteína mixta, involucrando regiones génicas E1 (primeros cuatro aminoácidos) y E4 (25,26). Forma complejos hexaméricos, capaces de unirse a la red de citoqueratina de la célula, a través de su región N-terminal produciendo su desestabilización. También produce alteración en el potencial de membrana mitocondrial. La región C-terminal permite su unión a la proteína del gen DEAD-box, alterando su función tipo helicasa, ATPasa, recambio RNA y estabilización de ribosomas (27). Su región intermedia (aminoácidos 17 a 45) detiene el ciclo celular en G2, a través de la unión a ciclinas y kinasas dependientes de ciclinas y por bloqueo de E7. Por estos mecanismos, la célula pierde soporte estructural por la alteración del citoesqueleto, produciendo daño mitocondrial y del metabolismo energético, además de apoptosis (26). Es posible que durante esta etapa de amplificación del genoma viral, la inhibición de las oncoproteínas E6 y E7, el daño al citoesqueleto, el trastorno mitocondrial-energético y la apoptosis permitirían una mayor diseminación de la progenie viral, siempre que las proteínas de la cápside viral (L1 y L2) hayan sido sintetizadas, formando con el ADN viral nuevos viriones infectantes.
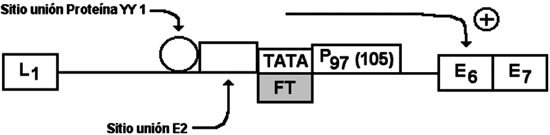
Un evento central para la transformación de las células infectadas es la integración del genoma viral al de la célula huésped, proceso que ocurre en el estrato espinoso y en el que destaca por su importancia, el ADN de los VPH de alto riesgo oncogénico. Durante este proceso se altera la región de lectura abierta ORF- E2, perdiéndose el efecto inhibitorio sobre el promotor P97 o P105, que mantenía bloqueada la expresión de las proteínas E6 y E7, las que son ahora sintetizadas sin restricción vía factores de transcripción (28) (Figura 3).
E6 es una proteína de 150 aminoácidos que liga Zinc; gran parte de sus funciones biológicas dependen de la integridad de 4 residuos de cisterna (29). Forma un complejo con p53 y la enzima ubiquitina ligasa, produciendo su degradación (30). El gen p53 se localiza en el cromosoma 17; la proteína p53 en condiciones basales, permite detener el ciclo celular en fase G1, efecto mediado por la proteína p21cip1/WAF1, que inhibe a las kinasas dependiente de ciclinas o activar el mecanismo de apoptosis, mediante la activación del gen bax, procesos desencadenados frente a una carga viral elevada o ante mutaciones del ADN celular. De este modo, su función protectora del genoma celular es alterada por la proteína E6. Otras funciones corresponden a la amplificación de la actividad telomerasa, inducir síntesis de ADN mutado y aumentar la integración del ADN viral al de la célula huésped (29).
El sistema proteína kinasa activada por mitógeno (MAP kinasa), vinculado a la proliferación celular, ha sido relacionado a la acción de oncoproteínas de VPH de alto riesgo (31,32). Entre sus componentes, Fosfatidilinositol-3-kinasa (PI3K), fosfatidilinositol bi y trifosfato (PIP2-PIP3), Rap-1, kinasa S6 y Akt, corresponden a sitios específicos de control, algunos de los cuales serán señalados a continuación.
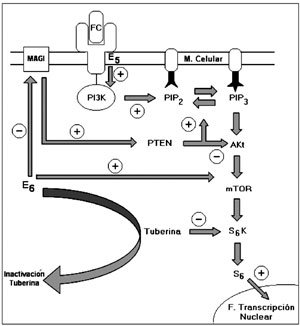
Se ha descrito recientemente una proteína llamada tuberina, de 200 Kda, generada a partir de un gen supresor de tumores conocido como complejo de la esclerosis tuberosa (TSC tipo 2). Tuberina controla la proliferación celular a través de la inhibición de la proteína kinasa S 6 (31). E6 se une a tuberina, inhibiendo su función; la interacción de E6 con Rap-1 permite activar el sistema kinasa MAP También se ha descrito la interacción de E6 con proteínas de membrana conocidas como homólogos de guanilato ciclasa (MAGI 2 y 3). Estas proteínas en condiciones normales estimulan a PTEN (homólogo de fosfatasa y tensina) el cual, en condiciones basales, controla el crecimiento celular inhibiendo el sistema, vía bloqueo de Akt y transformando PIP3 a PIP2 (31) (Figura 4). Al degradarse MAGI 2 y 3 por acción de E6 se perdería el efecto regulador del crecimiento celular.
E7 corresponde a una proteína de 100 aminoácidos, ligante de Zinc, que experimenta fosforilación, permitiéndole unirse a través de su extremo N-terminal (aminoácidos 20 a 30), a la proteína del retinoblastoma (pRB), alterando su función. PRB se origina del gen localizado en el cromosoma 13; interacciona con el factor de transcripción celular E2F en la fase G1 del ciclo celular, inhibiendo la expresión de genes relacionados a la replicación del ADN y proliferación celular (29). De esta forma, la unión de E7 con pRB, a través de la fosforilación de esta última, permite la expresión de timidina kinasa, c-myc, polimerasa a, PCNA, Ki-67, proteína de mantención de minicromosomas (MCM), p16, ciclina A y E (33).
Otras funciones descritas para la proteína E7 corresponden a la unión con Kinasa de Histona H1, lo que favorece la acción transformante celular (29). Además E7 inhibe a la proteína p16ink4A, la cual en condiciones basales frena la multiplicación celular (30) (Figura 5). También se menciona su efecto mutagénico, productor de aneuploidía y favorecedor de la integración del genoma viral al de la célula huésped (29). De este modo, las oncoproteínas E6 y E7 favorecen la proliferación e inmortalización de células con una mayor carga de ADN mutado.
E5 es una proteína sintetizada en el genoma viral localizándose principalmente en la membrana plasmática celular, interactuando con algunos factores como el factor de crecimiento epidérmico, factor de crecimiento derivado de plaquetas, factor p 185-neu y factor estimulante de colonias (34); de este modo podría estimular la acción de estos diferentes factores vinculados a la proliferación celular. Otro mecanismo atribuido a E5 sería la de activar la vía kinasa MAP, asociado a proliferación celular y diferenciación (34).
La integración del genoma viral al de la célula huésped no ocurre en todos los casos de cáncer cervical, pudiendo explicarse por mutaciones en zonas represoras como la región Ying- Yang (YYI), que mantendría la expresión continua de E6 y E7 o por la producción de ARN «quiméricos», más estables, permitiendo mayor síntesis de estas oncoproteínas (28).
Los procesos previamente descritos permiten la proliferación celular no controlada con una mayor carga de mutaciones. Para la transformación maligna se requiere además, la expresión (mutaciones) de un conjunto de genes encargados de permitir el desprendimiento de las células neoplásicas, la invasión a los tejidos vecinos, facilitar la angiogénesis, eludir la respuesta inmune del huésped, metástasis a distancia, entre otras funciones (29). En este sentido, existe evidencia de la participación de mutaciones en la región 3p14.2 (gen histidina frágil), inducida en parte por la integración del genoma viral (VPH 16), además de aquellas en las regiones cromosómicas 3p22.2, 11q22.1 y 11q23.3 que se han asociado a persistencia de lesiones intraepiteliales y/o progresión a cáncer cervical (35).
BIBLIOGRAFÍA
- Aynaud O, Bialasiewics A, Carmody C, Ferenczy A, Guillemotonia A, Hügel H, et al. Papillomaviruses in human pathology: epidemiology, pathogenesis and oncogenic role. In: Gross GE, Barrasso R. Human Papilloma Virus Infection. A Clinical Atlas. Berlin: Ullstein Mosby, 1997; 1-16.
- Longworth MS, Laimins LA. Pathogenesis of Human Papillomaviruses in Differentiating Ephitelia. Microbiol Mol Biol Rev 2004; 68(2): 362-72.
- Beutner KR, Tyring S. Human Papillomavirus. Am J Med 1997; 102(5A): 9-15.
- Frattini MG, Lim HB, Laimins LA. In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differentiation-dependent late expression. Proc Natl Acad Sci USA 1996; 93: 3062-7.
- Muñoz N, Bosch FX, Sanjosé S, Herrero R, Castellsagué X, Shah K, et al. Epidemiologic Classification of Human Papillomavirus Types Associated with Cervical Cancer. N Engl J Med 2003; 348: 518-27.
- Chow LT, Nasseri M, Wolinsky SM, Broker TR. Human Papillomavirus Types 6 and 11 mRNAs from genital Condylomata Acuminata. J Virol 1987; 61(8): 2581-8.
- Giroglou T, Florin L, Schäfer F, Streeck RE, Sapp M. Human Papillomavirus Infection Requires Cell Surface Heparan Sulfate. J Virol 2001; 75(3): 1565-70.
- Evander M, Fraser IH, Payne E, Qi YM, Hengst K, McMillan NA. Identification of the alpha 6 integrin as a candidate receptor for papillomaviruses. J Virol 1997; 71(3): 2449-56.
- Bousarghin L, Touzé A, Sizaret PY, Coursaget P. Human Papillomavirus Types 16, 31, and 58 Use Diffrerent Endocytosis Pathways To Enter Cells. J Virol 2003; 77: 3846-50.
- Kirchhausen T. Clathrin. Annu Rev Biochem 2000; 69: 699-727.
- Anderson RGW. Caveolae: Where incoming and outgoing messengers meet. Review. Proc Natl Acad Sci USA 1993; 90: 10909-13.
- Merle E, Rose RC, LeRoux L, Moroianu J. Nuclear Import of HPV 11 L1 Capsid Protein Is Mediated by Karyopherin _2_1 Heterodimers. J Cell Biochem 1999; 74: 628-37.
- Nelson LM, Rose RC, Moroianu J. Nuclear Import Strategies of High Risk HPV 16 L1 Major Capsid Protein. J Cell Biol Chem 2002; 277(26): 23958-64.
- Ruesch MN, Stubenrauch F, Laimins LA. Activation of Papillomavirus Late Gene Transcription and Genome Amplification upon Differentiation in Semisolid Medium Is Coincident with Expression of Involucrin and Trasglutaminase but Not Keratin-10. J Virol 1998; 72(6): 5016-24.
- Holmgren SC, Patterson NA, Ozbun MA, Lambert PF. The Minor Capsid Protein L2Contributes to Two Steps in the Human Papillomavirus Type 31 Life Cycle. J Virol 2005; 79(7): 3938-48.
- Del Vecchio AM, Romanczuk H, Howley PM, Baker CC. Transient Replication of Human Papillomavirus DNAs. J Virol 1992; 66(10): 5949-58.
- Ling Peh W, Middleton K, Christensen N, Nicholls P, Egawa K, Sotlar K, et al. Life Cycle Heterogeneity in Animal Models of Human Papillomavirus- Associated Disease. J Virol 2002; 76(20): 10401-16.
- Frattini MG, Laimins LM. Binding of the human papillomavirus E1 origen-recognition protein is regulated through complex formation with the E2 enhancer- binding protein. Proc Natl Acad Sci USA 1994; 91: 12398-02.
- Doorbar J, Elston RC, Napthine S, Raj K, Medcalf E, Jackson D, et al. The E1-E4 Protein of Human Papillomavirus Type 16 Associates with a Putative RNA Helicase through Sequences in Its C Terminus. J Virol 2000; 74(21): 10081-95.
- Conger KL, Liu J-S, Kuo S-R, Chow LT, Wang TS-F. Human Papillomavirus DNA Replication. J Biol Chem 1999; 274(5): 2696-705.
- Masterson PJ, Stanley MA, Lewis AP, Romanos MA. A C-Terminal Helicase Domain of the Human Papillomavirus E1 Protein Binds E2 and the DNA Polymerase _- Primase p68 Subunit. J Virol 1998; 72(9): 7407-19.
- Penrose KJ and MCBride AA. Proteasome- Mediated Degradation of the Papillomavirus E2-TA Protein Is Regulated Phosphorylation and Can Modulate Viral Genome Copy Number. J Virol 2000; 74(13): 6031-8.
- Desaintes C, Demeret C, Goyat S, Yaniv M, Thierry F. Expression of the papillomavirus E2 protein in HeLa cells leads to apoptosis. EMBO J 1997; 16(3): 504-14.
- Webster K, Parish J, Pandya M, Stern PL, Clarke AR, Gaston K. The Human Papillomavirus (HPV) 16 E2 Protein Induces Apoptosis in the Absence of Other HPV Proteins and via a p53- dependent Pathway. J Biol Chem 2000; 275(1): 87-94.
- Davy CE, Jackson DJ, Wang Q, Raj K, Masterson PJ, Fenner NF et al. Identification of a G2 Arrest Domain in the E1-E4 Protein of Human Papillomavirus Type 16. J Virol 2002; 76(19): 9806-18.
- Raj K, Berguerand S, Southern S, Doorbar J, Beard P. E1-E4 Protein of Human Papillomavirus Type 16 Associates with Mitochondria. J Virol 2004; 78(13): 7199-207.
- Wang Q, Griffin H, Southern S, Jackson D, Martin A, McIntosh P, et al. Functional Analysis of the Human Papillomavirus Type 16 E1-E4 Protein Provides a Mechanism for In Vivo and In Vitro Keratin Filament Reorganization. J Virol 2004; 78(2): 821-33.
- Bosch FX, Lorincz A, Muñoz N, Meijer CJLM, Shah KV. The causal relation between human papillomavirus and cervical cancer. J Clin Pathol 2002; 55: 244-65.
- Hausen H. Papillomaviruses Causing Cancer: Eva sion From Host-Cell Control in Early Events in Carcinogenesis. Review. JNCI 2000; 92(9): 690-8.
- Nakahara T, Nishimura A, Tanaka M, ueno T, Ishimoto A, Sakai H. Modulation of the Cell Division Cycle by Human Papillomavirus Type 18 E4. J Virol 2002; 76(21): 10914-20.
- Lu Z, Hu X, Li Y, Zheng L, Zhou Y, Jiang H, et al. Human Papillomavirus 16 E6 Oncoprotein Interferences with Insulin Signaling Pathway by Binding to Tuberin. J Biol Chem 2004; 279(34): 35664-370.
- Chakrabarti O, Veeraraghavalu K, Tergaonkar V, Liu Y, Androphy EJ, Stanley MA, et al. Human Papillomavirus tipe 16 E6 AminaAcid 83 Variants Enhance E6 Mediated MAPK Signaling and Differentially Regulate Tumorigenesis by Notch Signaling and Oncogenic Ras. J Virol 2004; 78(11): 5934-45.
- Middleton K, Peh W, Southern S, Griffin H, Sotlar K, Nakahara T, et al. Organization of Human Papillomavirus Productive Cycle during Neoplastic Progression Provides a Basis for Selection of Diagnostic Markers. J Virol 2003; 77(19): 10186-201.
- Gu Z, Matlashewski G. Effect of Human Papillomavirus Type 16 Oncogenes on MAP Kinase Activity. J Virol 1995; 69(12): 8051-6.
- ELhamidi A, Hamoudi RA, Kocjan G, Du M-Q. Cervical intraepithelial neoplasia: prognosis by combined LOH analysis of multiple loci. Gynecol Oncol 2004; 94: 671-9.


