2016
5
Autores:
Olivia Lafalla Bernad1, Manuel Domínguez Cajal2, Beatriz Obón Castel1, Leyre Ruiz Campo1, Mónica Hernández Aragón1, Cristina Abad Rubio1, Carlos Gómez González3, Ana Ma Vela Lete1
Instituciones:
1Servicio de Ginecología y Obstetricia., 2Unidad de Gastroenterología y Hepatología, 3Servicio de Anatomía Patológica. Hospital San Jorge. Huesca. España.
Imágen 1:
Imágen 2:
Imágen 3:
Tipo de documento:
Casos ClínicosDOLOR ABDOMINAL AGUDO COMO PRESENTACIÓN DE UN LINFOMA OVÁRICO
Contenido del documento:
RESUMEN
Antecedentes: El linfoma no Hodgkin de ovario es una patología infrecuente, más aun el ovárico primario. Caso Clínico: Presentamos el caso de una paciente con antecedente de diverticulitis aguda y varios episodios posteriores de dolor abdominal agudo, fiebre y aumento de reactantes de fase aguda con pruebas de imagen no concluyentes. A pesar de la buena respuesta inicial al tratamiento antibiótico, durante uno de los episodios la paciente precisa intervención quirúrgica urgente. Durante la intervención se halla una enfermedad tumoral avanzada con diagnóstico histológico de linfoma difuso de células grandes B. Conclusión: En el diagnóstico diferencial ante una sintomatología similar a la de nuestra paciente, se debería tener presente la posibilidad de un linfoma ovárico, a pesar de su baja incidencia.
PALABRAS CLAVE: Tumores ováricos, linfoma No Hodgkin
SUMMARY
Background: Non-Hodgkin´s lymphoma of the ovary is an infrequent pathology, even more primary one. Clinical case: We report a patient with a history of acute diverticulitis and several episodes of acute abdominal pain, fever and increased acute phase reactants with inconclusive imaging tests. Patient required urgent surgery during one episode despite a good initial response to antibiotic therapy. A locally advanced tumour disease was found in surgical intervention which was diagnosed as a diffuse large B-cell lymphoma. Conclusion: In the differential diagnosis in a manner similar to that of our patient symptoms, we should keep in mind the possibility of an ovarian lymphoma despite their low incidence.
KEY WORDS: Ovarian neoplasms, Non-Hodgkin lymphoma
INTRODUCCIÓN
El linfoma difuso de células grandes B (DLBCL) es el tipo más común de los Linfomas No Hodgkin (LNH) agresivos, que se suele presentar como una masa de crecimiento rápido acompañada hasta en un 30% de casos de síntomas B (fiebre, pérdida de peso o sudor nocturno). En un 40-50% de pacientes puede haber afectación extranodal secundaria, más frecuentemente en tracto gastrointestinal o en piel, y en un 10-35% de pacientes será extranodal primario (1). El ovario es una localización poco habitual del LNH, siendo más frecuente en el DLBCL y en el linfoma de Burkitt, describiéndose globalmente frecuencias del 10% en todo el tracto genitourinario. Habitualmente será invasión secundaria y menos frecuentemente podemos encontrar linfomas primarios ováricos, si bien la distinción entre primario y secundario puede ser compleja (2).
El objetivo de esta comunicación es presentar un infrecuente caso clínico de linfoma primario ovárico difuso de células grandes B.
Caso clínico
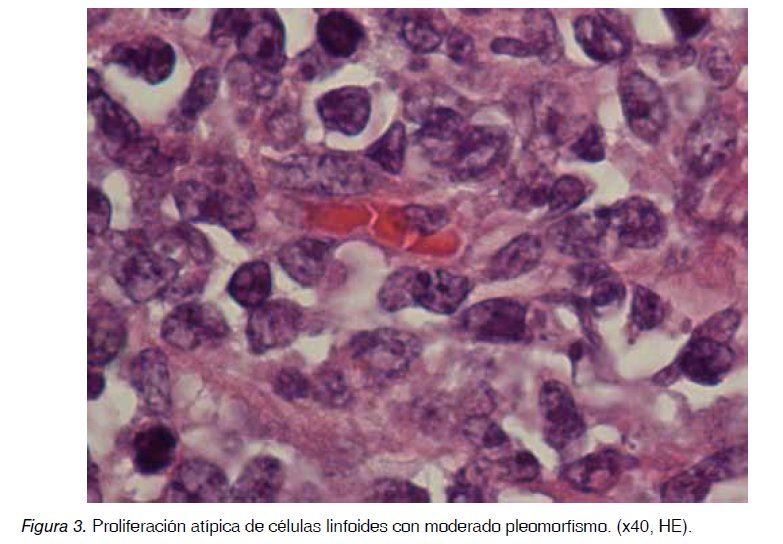
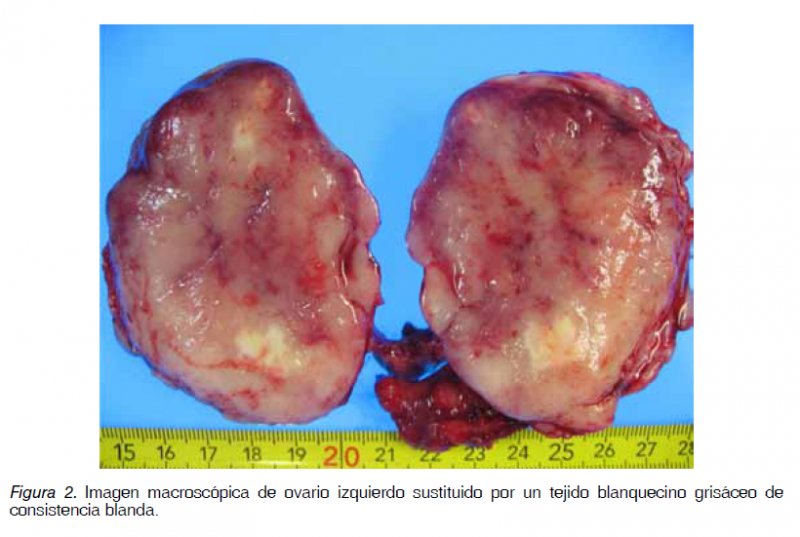
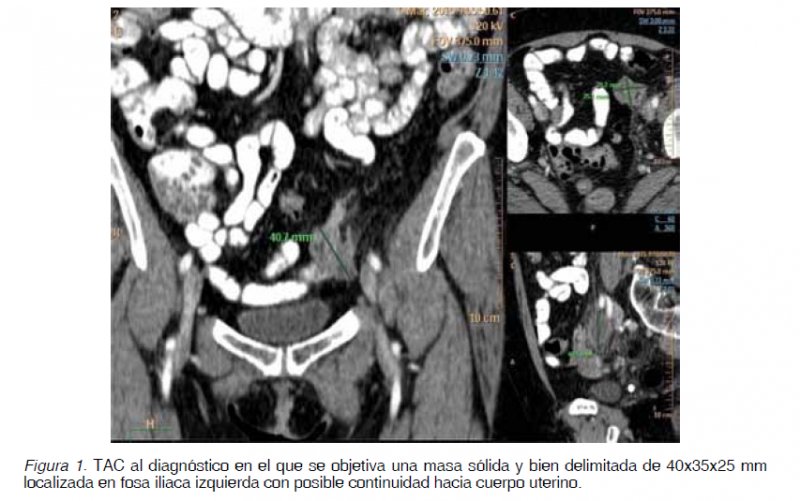
Mujer de 62 años con antecedente de un episodio de diverticulitis aguda confirmada radiológicamente y con una colonoscopia que confirmaba los divertículos y la ausencia de otras lesiones. A los 4 meses ingresa en la Unidad de Gastroenterología por un cuadro de dolor en fosa iliaca izquierda (FII) e hipogastrio de 24 horas de evolución acompañado de fiebre de 39ºC, sin asociar náuseas, vómitos, alteración del tránsito intestinal, ni otros datos de alarma. Analíticamente destacaba una leucocitosis con desviación izquierda (12.600/mm3 con 87% neutrófilos) y un aumento de reactantes de fase aguda (PCR: 48,8 mg/L; fibrinógeno: 440,38 mg/dL; VSG: 36 mm/1ª hora). Se realiza una tomografía axial computada (TAC) abdominal (Figura 1) en el que no se identifican complicaciones ni diverticulitis. En cambio se objetiva en FII una masa sólida y bien delimitada de 40x35x25 mm que parece presentar continuidad hacia el útero a través de una trompa dilatada. Además se visualiza cranealmente un nódulo de 24 mm sugestivo de adenopatía y otras cuatro adenopatías retroperitoneales de entre 11 y 15 mm. Como conclusión se informa que puede tratarse de un proceso inflamatorio tubo-ovárico. Por este motivo es valorada por el Servicio de Ginecología sin objetivarse alteraciones en la exploración, y en la ecografía transvaginal se visualiza un útero de tamaño y morfología regular con endometrio homogéneo de aspecto atrófico, ovario derecho de 17 mm sin lesiones y ovario izquierdo con una formación quística de 26×21 mm de contenido jaspeado, sin papilas ni tabiques. Se completa el estudio con marcadores tumorales (Ca 125, CEA, Ca 19.9 y HE-4) todos dentro de la normalidad. Durante el ingreso es tratada mediante antibioterapia de amplio espectro con buena respuesta por lo que se procede al alta pendiente de completar el estudio ambulatoriamente. A los 10 días reingresa por un cuadro similar aunque de menor intensidad por lo que se reinicia el antibiótico y se realiza una resonancia magnética (RM) abdomino-pélvica en la que se objetivan adenopatías hipercaptantes en cadena paraórtica izquierda y una masa hipercaptante de 25 mm de posible origen anexial izquierdo que asocia una colección quistoidea, planteándose el diagnóstico diferencial entre proceso infeccioso o tumor anexial. Tras finalizar el tratamiento antibiótico y descartando otros procesos infecciosos, incluyendo TBC, se decide realizar un nuevo TAC, que al igual que en la RM se objetiva una masa sólida polilobulada de 40x35x25 mm sospechosa de masa anexial izquierda y persisten las imágenes sugestivas de adenopatías. En nueva ecografía transvaginal se objetivan varias adenopatías de aspecto inflamatorio sin otras lesiones sospechosas. Ante la duda en el origen se realiza una punción percutánea guiada por TAC de la masa. La muestra es informada como tejido constituido por linfocitos, probablemente en relación con ganglio linfático y tejido fibroso moderadamente celular sin signos de malignidad. Ante estos hallazgos se decide mantener una actitud conservadora y valorar la evolución de las lesiones dado su posible origen inflamatorio. Sin embargo, a las tres semanas de la punción vuelve a presentar un cuadro de dolor, con mala evolución del mismo, por lo que tras un nuevo TAC urgente que no muestra cambios relevantes con respecto al previo, se decide realizar intervención quirúrgica urgente por parte de Ginecología. En la intervención se encuentra una masa sólida, dura, de unos 7 cm en ovario izquierdo, abrazada por la trompa de Falopio y en continuidad con adenopatías iliacas duras, de 2-3 cm que infiltran el recorrido en esa zona ureteral, alcanzando pared y retroperitoneo. No hay ascitis y útero y anejo derecho son normales. Se realiza una histerectomía total con anexectomía bilateral, exéresis de adenopatías retroperitoneal e iliaca, desestimando Urología la liberación ureteral por el riesgo de la técnica debido a la gran infiltración retroperitoneal. Posteriormente, el informe de Anatomía Patológica describe sobre la pieza de anexectomía izquierda una neoformación de estirpe linfoide de patrón difuso formando células grandes de hábito centroblástico, inmunoblástico y algunas células polimorfas, con expresividad para CD20, BCL2, BCL6, KI 67 y MUM 1, compatible con un linfoma no Hodgkin difuso de célula grande tipo B (no centrofolicular) variante citológica centroblástica polimorfa, con afectación de trompa de Falopio ipsilateral (Figuras 2, 3 y 4). Igualmente se objetiva afectación de los ganglios locales y retroperitoneales extirpados.
Para completar el diagnóstico se realiza un tomografía por emisión de positrones (PET) previo al inicio del protocolo quimioterápico (Figura 5), objetivándose múltiples adenopatías supra e infradiafragmáticas (nivel ganglionar IIb derecho, III bilateral, supraclavicular izquierda, retroperitoneales de hasta 8 cm, iliacas y pélvicas). Hematología instaura tratamiento con protocolo rituximab, ciclofosfamida, clorhidrato de doxorubicina (hidroxidaunorubicina), sulfato de vincristina (Oncovin) y prednisona (R-CHOP) con buena tolerancia y con respuesta completa en el PET tras completarlo.
DISCUSIÓN
El DLBCL es el tipo de linfoma más común, representando aproximadamente el 25% de todos los LNH y suponiendo en Europa una incidencia de casi 5 casos por cada 100.000 habitantes/año, aumentando ésta con la edad (mediana de 64 años al diagnóstico) (3). Se trata de un LNH agresivo que se suele presentar como una masa de crecimiento rápido, habitualmente secundario al aumento de tamaño de adenopatías del cuello o del abdomen. En el 30-40% de los casos, más comúnmente en los agresivos o con afectación extranodal, puede ir asociado a síntomas B, y en la mitad de los casos se objetivará un aumento de lactato deshidrogenasa (LDH). El 60% se diagnosticará en fases avanzadas y sólo el 40% estará localizado (estadios I y II de Lugano) (Tabla I). Puede haber afectación de médula ósea en el 30% y extranodal en el 40-50% de los pacientes durante su evolución natural, principalmente gastrointestinal y cutánea, si bien puede localizarse en cualquier tejido (4). Entre el 10-35% de los casos pueden presentarse como un linfoma primario extranodal (5).
El diagnóstico debe realizarse por estudio morfológico e inmunofenotipado de tejido ganglionar o de otros tejidos si no hay afectación nodal. Las células tumorales expresan antígenos pan-B (CD19, CD20, CD22, CD79a) por lo que suelen diagnosticarse mediante estas tinciones salvo en casos atípicos. Dado que las adenopatías son comunes en cuadros infecciosos e inflamatorios, la evaluación inicial debe ir enfocada a la exclusión de estas causas. Debería biopsiarse los ganglios en caso de tamaño significativo (>2 cm de diámetro o >1,5×1,5 cm), aumento o persistencia durante 4-6 semanas. Debido a una mayor facilidad es preferible la biopsia de ganglios periféricos pero en ocasiones debe realizarse guiada por TAC o por laparoscopia.
En la evaluación inicial debe establecerse el tipo histológico preciso, la extensión y el estado general del paciente para decidir el tratamiento. El pronóstico dependerá principalmente de la histopatología y se utilizará el Índice Pronóstico Internacional para los LNH que incluye la edad, el Performance Status, la LDH, el estadio y el grado de afectación extranodal.
En los estadios limitados el tratamiento aconsejado son 3 ciclos de quimioterapia con ciclofosfamida, doxorubicina, vincristina y prednisona, junto con el anticuerpo recombinante anti-CD20 Rituximab (R-CHOP) seguido de radioterapia. En los estadios avanzados el tratamiento de elección son 6-8 ciclos de R-CHOP cada 3 semanas. A pesar de los avances, gran parte de los pacientes presentarán recidiva o refractariedad, en cuyo caso se iniciará quimioterapia sistémica, con o sin Rituximab, y transplante de células hematopoyéticas en aquellos respondedores a la quimioterapia (6,7).
El ovario es una localización poco habitual de LNH, siendo más frecuente en el caso del DLBCL y del linfoma de Burkitt, describiéndose frecuencias del 10% globalmente en el tracto genitourinario. Habitualmente será una invasión secundaria que se diagnosticará durante el estudio de extensión. El linfoma primario ovárico es infrecuente (1,5% de los tumores ováricos y 0,5% de todos los LNH) por lo que únicamente hay datos de casos clínicos o series cortas de casos (8,9). En este caso se estadifica mediante el sistema de la Federación Internacional de Ginecología y Obstetricia (FIGO) (Tabla II).
No existen estudios clínicos que indiquen cual es el tratamiento más adecuado, si bien es práctica común la realización de salpingooforectomía bilateral a la que se puede añadir una histerectomía. También puede aceptarse la realización únicamente de ooforectomía unilateral si no existe ninguna evidencia de afectación de ambos ovarios. Siguiendo el estadio del DLBCL se añade quimioterapia adyuvante (R-CHOP) y en algunos centros se realiza profilaxis de afectación del Sistema Nervioso Central. A pesar de que el tratamiento y el pronóstico son diferentes en linfomas primarios o secundarios, su distinción suele ser dificultosa por lo que el tratamiento se ajusta individualmente (10).
CONCLUSIÓN
Nuestra paciente debutó con varios cuadros de dolor abdominal agudo asociados a fiebre y elevación de reactantes de fase aguda, que mejoraban con tratamiento antibiótico lo que dificultó tener un alto grado de sospecha inicial de patología oncológica. La punción de la masa no aportó un diagnóstico anatomopatológico de malignidad, por lo que se orientó el cuadro hacia un origen inflamatorio y únicamente se decidió la intervención quirúrgica urgente por presentar una mala evolución. Durante la cirugía los hallazgos sugerían una enfermedad tumoral agresiva y avanzada pero sin características específicas decidiéndose la histerectomía con doble anexectomía y extirpación de adenopatías con fines diagnósticos. Tras el diagnóstico histológico se revisaron las pruebas de imagen iniciales, que al mostrar afectación del ovario y adenopatías de forma concomitante impedían la diferenciación entre el linfoma primario y secundario. En el diagnóstico diferencial ante una sintomatología similar a la de nuestra paciente, se debería tener presente la posibilidad de un linfoma ovárico a pesar de su baja incidencia.
BIBLIOGRAFÍA
- van Leeuwen MT, Turner JJ, Joske DJ, Falster MO, Srasuebkul P, Meagher NS, et al. Lymphoid neoplasm incidence by WHO subtype in Australia 1982-2006. Int J Cancer 2014;135(9):2146-56.
- Yadav R, Balasundaram P, Mridha AR, Iyer VK, Mathur SR. Primary ovarian non-Hodgkin lymphoma: Diagnosis of two cases on fine needle aspiration cytology. Cytojournal 2016;13:2.
- Sant M, Allemani C, Tereanu C, De Angelis R, Capocaccia R, Visser O, Marcos-Gragera R, Maynadié M, Simonetti A, Lutz JM, Berrino F; HAEMACARE Working Group. Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood 2010;116(19):3724-34.
- Møller MB, Pedersen NT, Christensen BE. Diffuse large B-cell lymphoma: clinical implications of extranodal versus nodal presentation–a population-based study of 1575 cases. Br J Haematol 2004;124(2):151-9.
- Anderson T, Chabner BA, Young RC, Berard CW, Garvin AJ, Simon RM, DeVita VT Jr. Malignant lymphoma. 1. The histology and staging of 473 patients at the National Cancer Institute. Cancer 1982;50(1):2699-707.
- Pfreundschuh M, Trümper L, Osterborg A, Pettengell R, Trneny M, Imrie K, et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol 2006;7(5):379-91.
- Pettengell R, Linch D, Haemato-Oncology Task Force of the British Committee for Standards in Haematology. Position paper on the therapeutic use of rituximab in CD20-positive diffuse large B-cell non-Hodgkin’s lymphoma. Br J Haematol 2003;121(1):44-8.
- Ambulkar I, Nair R. Primary ovarian lymphoma: report of cases and review of literature. Leuk Lymphoma 2003;44(5):825-7.
- Senol T, Doger E, Kahramanoglu I, Geduk A, Kole E, Yucesoy I, Caliskan E. Five cases of non-hodgkin B-cell lymphoma of the ovary. Case Rep Obstet Gynecol 2014;2014:392758.
- Zhao XY, Hong XN, Cao JN, Leaw SJ, Guo Y, Li ZT, Chang JH. Clinical features and treatment outcomes of 14 cases of primary ovarian non-Hodgkin’s lymphoma: a single-center experience. Med Oncol 2011;28(4):1559-64.


