2015
6
Autores:
Johana Arango1, Julián Delgado.1, Wilmar Saldarriaga.1, 2
Instituciones:
1Departamento de Ginecología y Obstetricia, Universidad del Valle, Hospital Universitario del Valle “Evaristo García”
Imágen 1:

Imágen 2:
Imágen 3:
Tipo de documento:
Casos ClínicosESCLEROSIS TUBEROSA, DIAGNÓSTICO FETAL Y MATERNO
Contenido del documento:
RESUMEN
La esclerosis tuberosa (ET) es una enfermedad genética, autosómica dominante que tiene expresividad variable y que se caracteriza por la presencia de hamartomas en múltiples órganos de diferentes sistemas (piel, cerebro y corazón). Es causada por mutaciones en los genes TSC1 locus 9q34 y TSC2 locus 16p13. Tiene una prevalencia de 1 en cada 5000 a 10000 recién nacidos vivos. Se reporta el caso de una gestante y su feto con diagnóstico de ET. Se observaron masas en corazón y cerebro en el feto. El examen físico exhaustivo de la embarazada mostró criterios mayores de la enfermedad. Los hallazgos del diagnóstico prenatal fueron confirmados en el recién nacido. Se contribuye a la construcción de datos epidemiológicos latinoamericanos, y se aporta un plan de manejo a fetos, embarazadas y recién nacidos con ET.
PALABRAS CLAVE: Esclerosis tuberosa, rabdomiomas, angiomiolipomas, diagnóstico prenatal
SUMMARY
Tuberous Sclerosis (TS) is a genetic disorder, exhibits an autosomal dominant inheritance pattern with variable expression, characterized by the presence of hamartomas in multiple organs of various systems (skin, brain and hearth). It is caused by mutations in genes TSC1 loci 9q34 and TSC2 loci 16p13. It has a prevalence of 1 in every 5000 to 10000 live births. We report a case of a pregnant and her fetus with diag- nosis of TS. Masses in heart and brain in the fetus were observed, the thorough physical examination of pregnant also showed major criteria of the disease. Prenatal diagnosis findings were confirmed in the new- born. The objective is contribute to the construction of Latin American epidemiological data, a management plan for fetuses, pregnant women and infants with TS is provided.
KEY WORDS: Tuberous sclerosis, rhabdomyomas, angiomyolipoma, prenatal diagnosis
INTRODUCCIÓN
La esclerosis tuberosa (ET) es una enferme- dad genética que tiene expresividad variable, que se caracteriza por la presencia de hamartomas en diferentes órganos con mayor frecuencia ubicados en piel, cerebro y corazón. El fenotipo incluye re- tardo mental, convulsiones y autismo (1). Tiene un patrón de herencia autosómico dominante, causa- da por mutaciones en los genes TSC1 locus 9q34 y TSC2 locus 16p13 (2), que son genes supresores tumorales que codifican las proteínas hamartina y tuberina, respectivamente, estas están involucra- das en la regulación de la proliferación celular (3). La ET es una enfermedad poco frecuente con una prevalencia de 1 de cada 5000 a 10000 na- cidos vivos, y de estos, dos tercios de los casos se presentan de forma esporádica (4). En Latino América no existen datos de prevalencia. El diagnóstico prenatal de ET se basaprincipalmente en la observación de tumores cardíacos múltiples (5), hasta un 88% de los fetos con rabdomiomas tienen la enfermedad (6). Otro órgano afectado con frecuencia por la ET en el feto es el cerebro, los hallazgos sugestivos son tubérculos corticales y/o nódulos sub ependimarios. En estos casos se deben buscar signos clínicos de ET en los padres. El objetivo de esta comunicación es reportar el caso de una gestante y su feto con diagnóstico de ET. La sospecha clínica de la patología en la emba- razada fue secundaria a los hallazgos de lesiones cardiacas y cerebrales en el feto. Se contribuye a la construcción de datos epidemiológicos latinoa- mericanos, y aportar un enfoque de estudio y de manejo sugerido por los autores al gineco-obstetra Latinoamericano.
Caso clínico
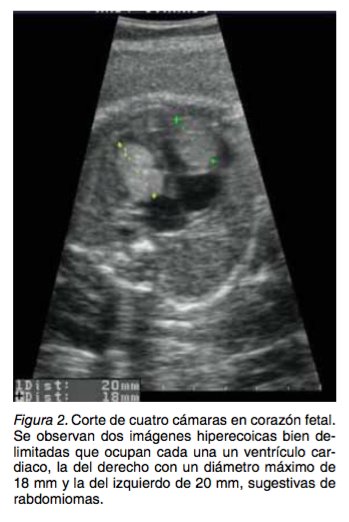
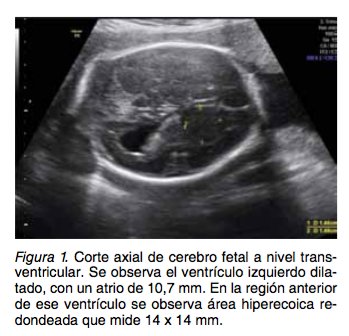
Paciente de 27 años, Gesta 2, Para 1, Mortinato 1, con 28 semanas de edad gestacional, ingresa al servicio de perinatología del Hospital Universitario del Valle, un hospital nivel III-IV que atiende pobla- ción pobre en su mayoría del régimen subsidiado del Suroccidente Colombiano. Es remitida por ha- llazgo de ventriculomegalia cerebral en ecografía obstétrica básica. En la ecografía nivel III, en el cor- te axial del cerebro fetal se observa imagen suges- tiva de un nódulo sólido (Figura 1). Además, en esa ecografía y en ecocardiograma fetal se reportaron dos imágenes sugestivas de rabdomiomas (Figura 2); no se observa obstrucción de los tratos de en- trada o salida cardiacos al momento del examen. Con los hallazgos ecográficos se sospecha escle- rosis tuberosa en el feto. El único antecedente médico y obstétrico en contrado fue un parto de pretémino a las 32 sema- nas de gestación, se obtuvo un mortinato de causa no clara, 18 meses antes de este embarazo, no pudiendo concluirse que estuviese relacionado con ET.
Una semana después, la gestante con 29 se- manas es hospitalizada por amenaza de parto pretérmino. Durante la hospitalización es llamati- vo al examen físico de la madre múltiples pápulas cupuliformes en mejillas, pared nasal, en zonas adyacentes a surcos nasogenianos. Es evaluada por dermatología que concluyen que esas lesiones son angiofibromas faciales (Figura 3). Además, en- cuentran maculas hipopigmentadas en miembros inferiores (Figura 4) y parche lijoso en el dorso, paravertebral a altura de T8 (Figura 5). En ecogra- fía renal y de vías urinarias se evidencian lesiones renales múltiples y bilaterales sugestivas de an- giomiolipomas, al menos 4 en el riñón derecho, de hasta 4 centímetros de diámetro, y 2 lesiones en el riñón izquierdo de 1 y 2,5 centímetros. La valo- ración por oftalmología no mostró hallazgos signi- ficativos. Por la sumatoria de los hallazgos en la gestante y el feto se hizo diagnóstico de ET.
Los síntomas de amenaza de parto prematuro fueron controlados, se dio alta con seguimiento en consulta externa como un embarazo de alto riesgo. La vigilancia del feto se efectuó con ecografías obs- tétricas especializadas observando específicamen- te si las masas cardiacas producían obstrucción en los tractos de salida o entrada al corazón, y si había progresión de la ventrículomegalia cerebral con au- mento importante del diámetro biparietal o del perí- metro cefálico. Ningún signo de complicación fetal fue encontrado; se realizó cesárea a la semana 38, por los hallazgos renales que contraindicaban el parto vaginal.
Se recibió un recién nacido masculino de 2496 gramos, sin complicaciones. Se traslada el neonato a la unidad de cuidado intensivo neonatal, donde
se realiza ecocardiograma que evidencia masas intracardiacas, sin afectación de funcionalidad, sin repercusión hemodinámica, y sin hipertensión pul- monar. La valoración por oftalmología no mostró hallazgos patológicos. La resonancia magnética nuclear (RMN) cerebral mostró una lesión intra- ventricular a nivel del cuerno frontal del ventrículo izquierdo de 3,5 x 2,5 cm, con dilatación del siste- ma ventricular generalizado, predominante a nivel supratentorial, también se observan nódulos en el ventrículo derecho menores de 10 mm de diámetro. Los servicios de neurocirugía y neurología pediá- trica concluyeron que el recién nacido no requería de procedimientos invasivos intracerebrales dada la tendencia a la regresión de las masas. No se en- contraron hallazgos dermatológicos. Se da egreso a la madre y su hijo con control ambulatorio. Al mo- mento del sometimiento de este artículo la mujer y su hijo estaban en buenas condiciones. La madre firma consentimiento informado aceptando la toma de fotografías y uso de la información de la historia clínica de ella y su hijo para esta publicación.
DISCUSIÓN
La ET es una patología con un origen genético con un patrón de herencia autosómico dominante que cumple con las definiciones de expresividad variable y pleiotropía, dado que puede afectar mul- tiples órganos, no siendo constante el fenotipo en los afectados (7). Dada la ubicación y tamaño de los hamartomas los pacientes pueden presentar múltiples síntomas, entre ellos convulsiones, autis- mo, pérdida visual, falla renal, arritmias cardiacas entre otros (1,8,9).
Los análisis genéticos han mostrado mutacio- nes en los genes TSC1 y TSC2, lo cual produce alteración en la migración y diferenciación de las células de la cresta neural, que a su vez tienen un origen ectodérmico y migran a múltiples órganos de otro diferente origen embriológico (10). Esa carac- terística, lleva al desarrollo de tumores benignos, circunscritos, no invasivos en órganos de diferen- tes sistemas. Las lesiones son de tipo angiomioli- pomas y en células epiteliales perivasculares (11).
La amplia variación en el alcance de la ubica- ción de la migración de las células de la cresta neu- ral y la gravedad de las manifestaciones clínicas, muestra que no existe correlación estricta entre una mutación y sus resultados clínicos, llevando a la expresividad variable y la pleiotropía de la en- fermedad, inclusive en los casos heredados, como en el aquí reportado no hay consistencia en los hallazgos fenotípicos madre e hijo. Sin embargo, se ha podido documentar que pacientes con muta- ciones en TSC2 tienen síntomas más severos que aquellos con mutaciones en TSC1, presentando tubérculos corticales con epilepsia, retraso mental
(moderado y severo), angiomiolipomas renales, hamartomas de la retina, y angiofibromas faciales. Además, otros estudios, han mostrado que cuando hay asociación entre un alelo de alta expresión del gen que codifica el interferón gamma y una baja frecuencia de angiomiolipomas renales en pacien- tes con mutaciones en TSC2, sugiriendo esto que existen genes modificadores que pueden generar efectos importantes en el fenotipo (8).
El diagnóstico prenatal se basa generalmente en hallazgos de tumores cardíacos múltiples (12). Series de casos reportados en la literatura han mostrado que ante la evidencia de rabdomiomas cardiacos, hasta un 88% de los fetos pueden pre- sentar diagnóstico ET, a su vez, aproximadamente en el 30% los fetos con ET tendrán rabdomiomas (6). Se debe tener en cuenta, que este tipo de lesio- nes cardiacas van a tener una tendencia a la invo- lución espontánea en la vida extrauterina, pero que in útero pueden crecer a lo largo de la gestación y producir obstrucción de los tractos de entrada y salida del corazón y llevar a la muerte del feto (13).
El diagnostico prenatal, exige una valoracion ecografica detallada de otros órganos, como ocu- rrió en el caso reportado (14). A nivel cerebral en fetos con ET se pueden observar tubérculos cor- ticales, nódulos subependimarios y astrocitomas subependimarios de células gigantes (8), que se encuentran en el 90% de los casos; si bien, estos se forman durante las 7-12 semanas de gestación, pueden ser evidentes entre las 20-26 semanas, en ecografía o por RMN (15).
En el abdomen en el diagnóstico prenatal, se pueden encontrar masas renales como angiomio- lipomas y quistes, y hamartomas hepáticos, con menor frecuencia que las anomalías cardiacas y cerebrales. Son significativamente más frecuentes y numerosas en pacientes con TSC2 que en los TSC1 (7).
Todo recién nacido que in útero se sospechó ET, debe tener RMN cerebral, electroencefalogra- ma, ecocardiograma, electrocardiograma, ecogra- fía renal y de hígado en los 5 primeros días de vida, además debe recibir valoración por neurología, of- talmología, nefrología, dermatología, neurocirugía y genetista para buscar otros signos de ET para precisar el diagnóstico (16).
Las embarazadas, padres y hermanos, de fetos y/o recién nacidos con sospecha de ET, deben ser valorados por genetistas clínicos para buscar sig- nos clínicos de la enfermedad, construir heredogra- mas y hacer la inferencia de si el caso es heredado o de novo (17). El diagnóstico de ET se realiza con dos criterios mayores, o uno mayor y dos menores (Tabla I). En el caso de la paciente y el recién na- cido, ambos cumplían criterios para hacer el diag- nóstico clínico.
En principio, los hallazgos sugestivos y relati- vamente fáciles de encontrar son los de piel, como en el caso aquí reportado, donde la embarazada tenía angiofibromas faciales (Figura 3), maculas hipopigmentadas en miembros inferiores (Figura 4) y parche lijoso (Figura 5). Estudios complementa- rios que se deben hacer en los integrantes de la familia en los que se sospeche ET son: RMN ce- rebral y de columna, ecografía abdominal total que incluya una valoración detallada hepática y renal, radiografía de tórax, electrocadiograma, ecocardio- grama, evaluación dermatológica y oftalmológica (16). La consejería genética en los casos hereda- dos a los afectados por ET se hace explicando que tienen un 50% de probabilidad de tener hijos con la enfermedad y que no es posible predecir cual será el grado de expresión del fenotipo, el cual puede ser desde leve con solo hallazgos en piel hasta se- vero con discapacidad intelectual, convulsiones de difícil manejo, tumores en sistema nervioso central, corazón, riñones o hígado (18). Las pruebas moleculares que confirman del diagnóstico clínico son amplificación por PCR, se- guida de secuenciación directa y/o mediante MLPA Gestante X X X X Feto X X X (Multiplex Ligation-dependent Probe Amplification) o PCR cuantitativa en tiempo real (qPCR Real Time) para la detección de reordenamientos géni- cos en los genes TSC1 y 2. La mutación ha podido ser identificada en el 85% de los afectados. De es- tos en el 31% la alteración ha sido en el TSC1 y en 69% en el TSC2 (8). Dado que la alteración gené- tica es una mutación específica, en el diagnóstico prenatal o posnatal no se requiere estudios como cariotipo. Sobre el manejo de embazadas con ET, es poco lo que existe documentado. En la literatura se encuentran reportes de casos y sugerencias sustentadas en análisis fisiopatológicos. Así, se ha- cen recomendaciones puntuales sobre hallazgos específicos; cuando se diagnostican angiolipomas renales mayores de 4 cm de diámetro, como en el caso de la paciente reportada, los cambios de presión arterial en el trabajo de parto y la valsal- va, podrían aumentar el riesgo de sangrado de los tumores renales, por lo cual se sugiere que la vía de finalización sea cesárea (19). Toda embarazada con ET antes del parto debe tener una RNM cere- bral y de columna en la que se descarten lesiones que ocupen espacio y o lesiones vasculares que contraindiquen la analgesia peridural o la aneste- sia raquídea, específicamente en las pacientes en las que se haya sumado el diagnóstico de epilepsia se deben sospechar lesiones del SNC, si no tienen RMN tendrían contraindicación para analgesia epi- dural o anestesia raquídea (20).
CONCLUSIÓN
El obstetra debe sospechar el diagnóstico de ET en fetos en los que se observan masas car- diacas, cerebrales corticales o intraventriculares. Las embarazadas con fetos con estos hallazgos se les debe realizar una historia clínica y examen físico exhaustivo buscando signos clínicos de ET, especialmente en piel. La evaluación puede ser complementada con RMN cerebral, ecocardiogra- ma, ecografía renal y de hígado. En casos en que embarazadas y fetos cumplan con criterios clínicos de ET se deben seguir recomendaciones específi- cas para el manejo obstétrico y neonatal, e incluir la consejería genética.
BIBLIOGRAFÍA
1. Roach ES, Gomez MR, Northrup H. Tuberous sclero- sis complex Consensus Conference: revised clinical diagnostic criteria. J Child Neurol 1998;13(12):624-8.
2. Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients 7.
indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet 2001;68(1):64-80.
3. Lee K, Won H, Shim JY, Lee P, Kim A. Molecular ge- netic, cardiac and neurodevelopmental findings in ca- ses of prenatally diagnosed rhabdomyoma associated with tuberoues sclerosis complex. Ultrasound Obstet Gynecol 2103;41(3):306-11.
4. Hallett L, Foster T, Liu Z, et al. Burden of disease and unmet needs in tuberous sclerosis complex with neu- rological manifestations: systematic review. Curr Med Res Opin 2011;27(8):1571-83.
5. Gedikbasi A, Oztarhan K, Ulker V, et al. Prenatal so- nographic diagnosis of tuberous sclerosis complex. J Clin Ultrasound 2011;39(7):427-30.
6. Yinon Y, Chiltayat D, Blaser S, et al. Fetal cardiac tu- mors: a single-center experience of 40 cases. Prenat Diagn 2010;30(10):941-9.
7. Borkowsk J, Schwartz R, Kotulska K, Jozwiak S. Tu- berous sclerosis complex: tumors and tumorigenesis. Int J Dermatol 2011;50(1):13-20.
8. Curatolo P, Bombardieri R, Jozwiak S. Tuberous scle- rosis. Lancet 2008;372(9639):657-68. 9. Chen CP, Su YN, Hung CC, et al. Molecular gene- tic analysis of the TSC genes in two families with prenatally diagnosed rhabdomyomas. Prenat Diagn 2005;25:176-9.
10. Jozwiak J, Jozwiak S, Wlodarski P. Possible mecha- nisms of disease development in tuberous sclerosis. Lancet Oncol 2008;9(1):73-9.
11. Glass T, Smith P, Hodges R, Holmes HJ. Intramural pregnancy presenting in a patient with tuberous scle- rosis. J Clin Ultrasound 2010;38(7):393-6.
12. Gedikbasi A, Oztarhan K, Ulker V, et al. Prenatal so- nographic diagnosis of tuberous sclerosis complex. J Clin Ultrasound 2011;39(7):427-30.
13. Knight CJ, Pembridge JM. Cardiac rhabdomyo- ma presenting with fetal bradycardia: is immedia- te delivery always the answer? J Obstet Gynaecol 2012;32(4):399.
14. Zhang XY, Meng H, Zhong DR, Jiang YX. Cardiac rhabdomyoma and renal cyst in a fetus: early onset of tuberous sclerosis with renal cystic disease. J Ultra- sound Med 2008;27(6):979-82.
15. Baskin HJ Jr. The pathogenesis and imaging of the tuberous sclerosis complex. Pediatr Radiol 2008;38(9):936-52.
16. Roach ES, DiMario FJ, Kandt RS, Northrup H. Tube- rous Sclerosis Consensus Conference: recommen- dations for diagnostic evaluation. National Tuberous Sclerosis Association. J Child Neurol 1999;14(6):401-
17. Roach ES, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol 2004;19(9):643-9.
18. Jones K, Jones M, del Campo M. Tuberous sclerosis syndrome. In: Smith’s Recognizable Patterns of Hu- man Malformation. Philadelphia: ELSEVIER; 7th Edi- tion, 2013. pp 660-1.
19. Illescas T, Montalvo J, Contreras E, et al. Angiomioli- pomas, esclerosis tuberosa y gestación. Ginecol Obs- tet Mex 2009;77(8):380-6.
20. Causse-Mariscal A, Palot M, Visseaux H, et al. Labor analgesia and cesarean section in women affected by tuberous sclerosis: report of two cases. Int J Obst Anest 2007;16(3):277-80.


