2012
1
Autores:
Marcelo Rodríguez G., Gabriela Egaña U.2, Rolando Márquez A., Maritza Bachmann M., Alejandro Soto A.
Imágen 1:
Imágen 2:

Tipo de documento:
DocumentosPREECLAMPSIA: MEDIADORES MOLECULARES DEL DAÑO PLACENTARIO
Contenido del documento:
RESUMEN
La preeclampsia se constituye como una de las complicaciones más serias del embarazo y una causa importante de mortalidad materna y perinatal. Pese a ser una de las patologías del embarazo más estudiadas, muchas dudas e incertidumbre rondan aún acerca de su mecanismo fisiopatológico. En esta revisión se intenta hacer un análisis sobre las nuevas teorías acerca de la fisiopatología de la preeclampsia, dando énfasis en las distintas moléculas que transfieren el daño placentario hacia el territorio sistémico.
PALABRAS CLAVE: Preeclampsia, placenta, apoptosis, estrés oxidativo
SUMMARY
Preeclampsia is one of the most serious gestational disease which causes maternal and perinatal mortality. In spite of being one of the most studied pathologies of the pregnancy there are many concerns around its physiopathology. In this review we attempt to analyze the new preeclampsia physiopathology theories, giving emphasis in the different molecules that transfer the placental damage towards the systemic territory.
KEY WORDS: Preeclampsia, placenta, apoptosis, oxidative stress
INTRODUCCIÓN
La preeclampsia corresponde a un defecto multisistémico definido por la presencia de hipertensión y proteinuria >300 mg/24 horas, en embarazos de más de 20 semanas(1-3). Su importancia radica en que constituye la primera causa de muerte materna en países en desarrollo (4,5), y pese a los múltiples avances en medicina, aún no existen estrategias eficientes para prevenir esta patología, siendo la interrupción del embarazo el único tratamiento efectivo actualmente disponible.
El objetivo de esta comunicación es revisar los mediadores moleculares involucrados en el daño placentario en preeclampsia.
FISIOPATOLOGÍA
Durante muchos años se ha considerado a esta patología como la enfermedad de las teorías, esto porque las múltiples hipótesis no lograban explicar el cuadro en su totalidad. Sin embargo, actualmente se ha propuesto una estructura fisiopatológica compuesta por 2 etapas (6,7)
Primera etapa o de Injuria Placentaria.
Ocurre durante las 20 primeras semanas de gestación, en la cual se produciría el fenómeno de placentación anómala (8). En este proceso se produce una invasión defectuosa del trofoblasto extravellositario, por lo cual las arterias espirales no experimentarían los cambios específicos del embarazo, esto es no habría reemplazo de las capas endoteliales y media por trofoblasto invasor, pérdida de elasticidad de la pared y del control vasomotor (9,10). De esta manera, el lecho vascular placentario no se transforma en territorio de baja resistencia, permaneciendo en un territorio de alta resistencia al flujo sanguíneo. Esto finalmente se traduce en un estado de vasoespasmo e isquemia local, lo que determina hipoxia y daño placentario (11-13).
Segunda etapa o de Disfunción Endotelial e Inflamación Sistémica
Disfunción endotelial: corresponde a la alteración de la función endotelial, caracterizada por un aumento de la concentración de agentes vasopresores y agregantes plaquetarios (endotelina 1 y TBX A2), y una disminución de las sustancias vasodilatadoras y antiagregantes plaquetarias (NO y PG2) (14,15). Este desbalance de sustancias vasoactivas, junto a una mayor sensibilidad a la angiotensina II, determinan un estado de vasoconstricción, produciéndose un aumento de la resistencia vascular periférica, y así un aumento de la presión arterial (16). A esto se asocia una mayor permeabilidad endotelial. Distintos estudios han demostrado un aumento de marcadores de disfunción endotelial en pacientes con preeclampsia en comparación con gestaciones normales, tales como aumento de relación PAI 1/PAI 2, factor von Willebrand, trombomodulina y VCAM-1 (17,18).
Inflamación sistémica: El embarazo normal se caracteriza por ser un estado de inflamación sistémica, en el cual hay una activación de monocitos, granulocitos y plaquetas (19). Además existe una mayor concentración plasmática de citoquinas proinflamatorias (20). En la preeclampsia existiría una respuesta inflamatoria sistémica (RIS) más exagerada, por lo que distintos marcadores de actividad proinflamatoria, tales como receptores de superficie, TNFα, IL-1ß e IL-6, se encuentran elevados (21-23). En este sentido, la disfunción endotelial sería parte de esta inflamación sistémica, constituyéndose en el elemento central de ésta.
De esta manera podemos distinguir en la preeclampsia 2 etapas. Una primera etapa asintomática, local, en la cual hay un estado hipóxico de la placenta, lo que determina una injuria de la misma, y una segunda etapa, sintomática, caracterizada por una RIS exagerada y una disfunción endotelial.
Entre estas 2 etapas existen mediadores, entendidos como moléculas o sustancias producidas por la placenta, y que son capaces de difundir este daño placentario y traducirlo en un compromiso sistémico (24-26). Diversos compuestos se han propuesto como mediadores, sin embargo, actualmente la evidencia destaca a 3 moléculas que han demostrado participar activamente en la fisiopatología de la preeclampsia, tanto en modelos in vitro como in vivo, y que son los que han generado las principales líneas de investigación. Éstos son los derivados del estrés oxidativo, los microfragmentos de sinciciotrofoblasto derivados de la apoptosis y los factores antiangiogénicos.
Estrés oxidativo: Corresponde al desbalance entre la producción de radicales libres y las sustancias antioxidantes (27). En este estado se favorece la generación de sustancias reactivas, entre las que destacan las sustancias oxígeno reactivas (ROS). Estas moléculas se caracterizan por tener 1 ó más electrones desapareados, por lo que son altamente inestables, de esta manera interactúan con fosfolípidos de membrana y ácidos grasos, formando así los lipoperóxidos, que son los encargados de generar el daño tisular (27).
En la placenta isquémica, secundario a una invasión defectuosa del trofoblasto, se produce una situación de hipoperfusión/reperfusión, favoreciendo un estado alternante entre hipoxia y normoxia (28). Este fenómeno es el gatillante de la activación de enzimas que generan ROS, tales como la NADPH y xantino oxidasas (29). A esto se asocia una disminución relativa de sustancias antioxidantes como superóxido dismutasa y vitaminas C y E (30). Este cuadro en conjunto determina que en pacientes con preeclampsia exista un aumento significativo de lipoperóxidos como malondialdehido (MDA) y F2 isoprostano (F2 ISO), en comparación a pacientes con gestación normal (31,32).
El daño en la preeclampsia sería a través de la transferencia de este estrés oxidativo placentario a la circulación materna vía lipoperóxidos, y también mediante la activación de neutrófilos que secretarían citoquinas proinflamatorias (33,34). El estrés oxidativo y la inflamación son procesos estrechamente relacionados, en el sentido que ambos son causa y a la vez consecuencia del otro; el estrés oxidativo produce inflamación sistémica y ésta a su vez es capaz de generar ROS localmente (24).
Apoptosis. Corresponde al proceso de muerte celular programada y que se describió por primera vez en 1972, cuando patólogos observaron un patrón de muerte celular en el cual los organelos experimentaron fragmentación, condensación y empaquetamiento con las membranas celulares dentro de estructuras densas, denominadas cuerpos apoptoicos, para luego ser fagocitados por células vecinas o macrófagos (35). De esta manera, no hay permanencia de restos celulares en el espacio extracelular, evitando así una respuesta inmune (36). A diferencia de la necrosis, la apoptosis es un fenómeno dependiente de energía celular.
Este proceso ocurre normalmente en todos los tejidos que están en permanente renovación, así también se constituye en parte importante del desarrollo embriológico de todos los órganos, incluida la placenta. En este sentido, el trofoblasto, dada su continua proliferación y renovación, presenta una tasa constante de apoptosis, que se inicia con la fusión sincicial y culmina con la extrusión del material apoptótico desde el sinciciotrofoblasto (STB) (37).
Los desechos subcelulares de este proceso se liberan a la circulación materna envueltas por una membrana protectora, impidiendo así el contacto con tejido materno y de paso evitar que estas estructuras induzcan una respuesta inflamatoria, permitiendo que sólo generen una pequeña activación de células inflamatorias maternas durante el embarazo normal (38). Este material apoptótico se puede encontrar en la sangre de las venas uterinas, y de ahí se dirigen al pulmón, en donde es digerida por macrófagos pulmonares, reduciendo sustancialmente la concentración de cuerpos apoptóticos en la sangre periférica de la madre (39). Para que este proceso ocurra exitosamente, se requiere de un período de tiempo necesario para que las distintas etapas se acoplen adecuadamente, lo que en general toma 3 a 4 semanas (40).
En la preeclampsia, en cambio, hay un incremento en la proliferación y fusión sincicial, por lo que la maquinaria para el proceso de degradación subcelular se encuentra sobreexigida. De esta manera, un mayor flujo de material celular no permite un tiempo suficiente para que la cascada apoptótica esté completa, antes de que ésta sea vertida en el torrente sanguíneo de la madre. Esto conduce a que partes necróticas del STB se desprendan y así liberan componentes intracelulares parcialmente degradados, sin una cubierta de membrana, los que en conjunto se denominan microfragmentos de sinciciotrofoblasto (STBM) (41,42).
Este desprendimiento necrótico de fragmentos intracelulares del sinciciotrofoblasto, incompletamente degradados, se conoce como aponecrosis, y se refiere a la disrupción de un proceso programado dependiente de energía (apoptosis) en favor de un proceso caótico independiente de energía (necrosis), que permite el desprendimiento de contenido celular, incluidas enzimas activas, dentro de un compartimiento donde produce daño tisular (43).
Los estados de hipoxia placentaria se asocian con aumento de la apoptosis a nivel del trofoblasto vellositario (44-46). Esto se correlaciona con el hallazgo de que en pacientes con preeclampsia se ha documentado una mayor concentración plasmática de STBM, en comparación a pacientes con embarazo normal (41). Estos STBM han demostrado in vitro ser capaces de alterar el comportamiento de macrófagos, y su aumento produce activación de neutrófilos, disrupción de células endoteliales y disminución de la relajación endotelial. De esta manera se pueden explicar tanto el daño placentario local (aumento del depósito de fibrinoide en la superficie de las vellosidades) como el daño sistémico y las manifestaciones clínicas de la preeclampsia (47-49).
Factores antiangiogénicos. Corresponden a proteínas que se unen directamente a factores de angiogénesis, impidiendo su unión con receptores de membrana en células endoteliales, propiciando la disfunción del endotelio (50). Se han descrito 2 factores:
1) Sflt-1: es una proteína antiangiogénica circulante, secretada principalmente por la placenta, y que corresponde a la forma truncada del receptor 1 del VEGF (VEGF R1 o flt-1) (51,52). Sólo está compuesta por el dominio extracelular del receptor, sin los dominios transmembrana ni intracelular, por lo que no es capaz de generar señales intracelulares. Debido a que es secretada hacia el plasma materno, es proclive a unirse a VEGF y PGF (factor de crecimiento placentario), evitando que éstos interactúen con sus receptores en el endotelio, no pudiendo así ejercer sus roles angiogénico y vasodilatador (53).
Su concentración plasmática se encuentra elevada en pacientes con preeclampsia, incluso desde estadíos preclínicos, aproximadamente 6 a 8 semanas antes del inicio de los síntomas (54). En estas pacientes, los niveles plasmáticos de sflt-1 se correlacionan con la intensidad de la enfermedad, y la caída de la concentración plasmática en el posparto a su vez se correlaciona con la mejoría clínica(55,56).
Estudios in vitro han demostrado que la disfunción endotelial inducida por plasma proveniente de pacientes con preeclampsia es revertida con la remoción del sflt-1 o con la adición de VEGF(57).
Modelos animales han corroborado su rol patogénico. Maynard y cols (56), en su estudio administraron sflt-1 a ratas preñadas y observaron que éstas presentaban un aumento significativo de la presión arterial y de la proteinuria. Además, evidenciaron que a nivel renal estas ratas presentaban las mismas lesiones histológicas características de las pacientes con preeclampsia, esto es la endoteliosis glomerular. Por otra parte, Li y cols (58), en su estudio demostraron que la adición de VEGF en ratas a las que previamente se les había administrado sflt-1, producía una disminución importante de la presión arterial y de la proteinuria, así como la mejora significativa de la histología renal (58). Esto último ciertamente que abre la esperanza de posibles opciones terapéuticas a investigar a futuro. Los estudios en humanos muestran resultados similares. Pacientes con cáncer tratados con proteínas antiangiogénicas que bloquean acción de VEGF (ac antiVEGF) para disminuir riesgo de metástasis, presentan como efectos secundarios aumento de la presión arterial y proteinuria (59).
2) s-eng: la endoglina es un co-receptor de superficie para TGF ß 1 y 3, el cual regula la vasodilatación dependiente de óxido nítrico (60). La forma soluble, o s-eng, corresponde a la forma truncada de la endoglina, e impide la unión de ésta al TGF ß, determinando de esta manera una disfunción endotelial (61). Es producida por la placenta, y su concentración plasmática se encuentra elevada en pacientes con preeclampsia (62).
En estudios in vivo también se ha documentado su efecto deletéreo sobre la función endotelial. La administración de s-eng a ratas preñadas demostró que éstas presentaban aumento de la presión arterial y proteinuria, aunque en menor rango que el producido por la administración de sflt-1 (63). Sin embargo, al administrar estos 2 factores antiangiogénicos en conjunto, se observó un efecto sinérgico de ambos, produciendo una proteinuria en rangos nefróticos, elevación de LDH y enzimas hepáticas, además de una disminución significativa del recuento de plaquetas, lográndose de esta manera producir un cuadro similar al observado en una preeclampsia severa y en el síndrome HELLP. De esta manera se postula que el desbalance angiogénico sería el elemento crucial en la patogénesis de la preeclampsia (64).
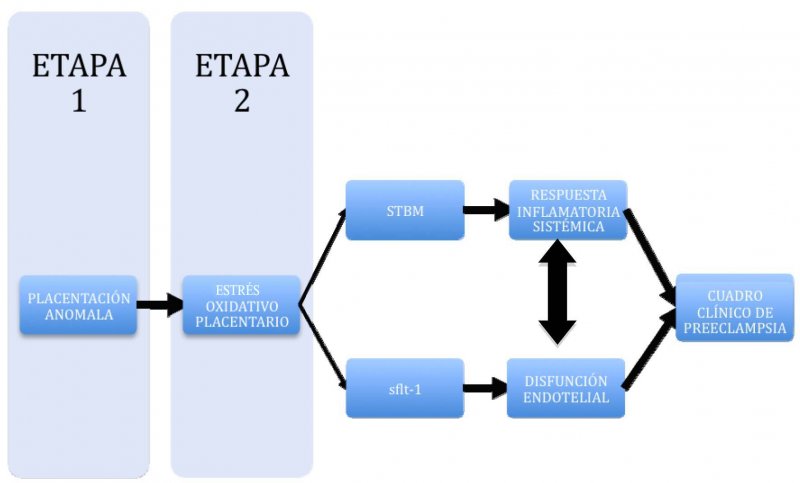
Tomando en cuenta toda esta evidencia en conjunto, Redman y Sargent (65), postularon en el 2005 una teoría fisiopatológica unitaria, que incorpora tanto a las 2 fases, como a los distintos mediadores recientemente descritos, participando de manera concertada y potenciando sus distintas acciones, para que en conjunto sean capaces de generar esta respuesta inflamatoria materna exagerada (Figura 1).
Si bien esta hipótesis es capaz de explicar aquel grupo de preeclampsias con alteración placentaria, que por lo general presentan un inicio precoz, con un curso severo, y asociadas en muchas ocasiones a restricción del crecimiento intrauterino (RCIU), no incluye a las pacientes con preeclampsia que no presentan alteración de la placentación, y que tienen un inicio más tardío, con un curso más benigno de la enfermedad y que logran muchas veces llegar hasta el final de la gestación (66). Este último grupo de pacientes muchas veces son obesas, asociadas a síndrome metabólico o a diabetes gestacional, cuadros que presentan basalmente una mayor concentración de los distintos marcadores de inflamación, tales como TNFα, IL-6, leptina y PAI-1(67); si a esta inflamación basal se adiciona un estado proinflamatorio como el embarazo normal, existe mayor riesgo de que esta respuesta inflamatoria global adquiera caracteres patológicos (68). Es en este sentido que las publicaciones más recientes plantean a la inflamación sistémica materna como el elemento fisiopatológico primario (67), sobre el cual actúan distintos situaciones que determinan una respuesta exagerada, como injuria placentaria, obesidad, síndrome metabólico, etc. (Figura 2).
¿PREECLAMPSIA COMO ENFERMEDAD ÚNICA?
Distintos autores plantean que la preeclampsia no es una enfermedad única, sino que tal vez se trate de diversas entidades patológicas que comparten en común un mismo criterio diagnóstico (69,70). Esto no sólo se basa en el sentido de que ninguna hipótesis fisiopatológica es capaz de explicar todos los cuadros diagnosticados como preeclampsia, sino que también por el hecho de presentar distintos factores epidemiológicos y pronósticos (71).
Las pacientes con preeclampsia severa (PES), que generalmente son de origen placentario, presentan mayor morbimortalidad cardiovascular a largo plazo que las pacientes con preeclampsia moderada (PEM) (72,73). La morbimortalidad perinatal en pacientes con PES está notablemente aumentada, sin embargo, en pacientes con PEM no se observa mayor morbimortalidad perinatal respecto de pacientes con embarazo normal (74). Esto queda de manifiesto en que no existen diferencias significativas en cuanto a complicaciones como desprendimiento placentario, parto prematuro, RCIU y mortalidad perinatal, incluso los recién nacidos de pacientes con PEM presentan mayor peso promedio que las pacientes con embarazo normal (75,76). Es en este sentido que los distintos autores proponen un cambio en la perspectiva de enfrentamiento de la preeclampsia como una enfermedad única, y comenzar a percibirla más bien como un síndrome.
CONCLUSIÓN
La preeclampsia corresponde a una de las complicaciones más importantes del embarazo, y pese a ser una entidad frecuente, se desconoce aún a cabalidad su fisiopatología. Sin embargo, en los últimos años se han hecho avances importantes en su comprensión, entre los cuales se menciona el hallazgo de mediadores bioquímicos que son capaces de traducir este daño primario placentario en un compromiso sistémico posterior. Por tal motivo, la comprensión precisa de estas moléculas patógenas y de su mecanismo de acción nos ayuda a entender de mejor manera a la preeclampsia desde el punto de vista fisiopatológico, y de paso nos da posibles blancos terapéuticos susceptibles de ser estudiados.
BIBLIOGRAFÍA
1. Report of the National High Blood Pressure Education Program. Working group report on high blood pressure in pregnancy. Am J Obstet Gynecol 2000;183;s1-22.
2. ACOG Practice Bulletin nº 33. Diagnosis and management of preeclampsia and eclampsia. Int J Gynecol Obstet 2002;77:67-75.
3. Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet 2005;365:785-99.
4. Duley L. The global impact of pre-eclampsia and eclampsia. Semin Perinatol 2009;33:130-137.
5. Donoso E. Mortalidad maternal en Chile, 2000-2004. Rev Chil Obstet Ginecol 2006;71:246-51.
6. Roberts J, Lain K. Recent Insights into the pathogenesis of pre-eclampsia. Placenta 2002;23:359-72.
7. Ness RB, Roberts JM. Heterogeneous causes constituting the single syndrome of preeclampsia: a hypothesis and its implications. Am J Obstet Gynecol 1996;175:65-70.
8. Chaddha V, Viero S, Huppertz B, Kingdom J. Developmental biology of the placenta and origins of placental insufficiency. Semin Fetal Neonatal Med 2004;9:357-69.
9. Kaufmann P, Black S, Huppertz B. Endovascular trophoblast invasion: implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol Reprod 2003;69:1-7.
10. Lyall F, Bulmer JN, Duffie E, Coustins F, et al. Human trophoblast invasion and spiral artery transformation: the rol pf PECAM-1 in normal pregnancy, preeclampsia, and fetal growth restriction. Am J Pathol 2001,158:1713-21.
11. Soleymanlou N, Jurisica I, Nevo O, Ietta F, et al. Molecular evidence of placental hipoxia in preeclampsia. J Clin Endocrinol Metab 2005;90:4299-308.
12. Huppertz B. Placental origins of preeclampsia: challenging the current hypothesis. Hypertension 2008;51:970-5.
13. Foidart J. Redistribution of the myometrial circulation in the uterus of women with preeclampsia or intrauterine growth retardation. Workshop1- Ninth Meeting of the International Federation of Placenta Associations, Mainz-Gissen, Germany, 2003.
14. Granger J, Alexander B, Llinas M, Bennett W, et al. Pathophysiology of preeclampsia: linking placental ischemia/hypoxia with microvascular dysfunction. Microcirculation 2002;9:147-160.
15. Poston L. Endothelial dysfunction in preeclampsia. Pharmacol Rep 2006;58 suppl:69-74.
16. Stepan H, Faber R, Dornhofer N, Huppertz B, et al. New insights into the biology of preeclampsia. Biol Reprod 2006;74:772-76.
17. Hubel CA, Lyall F, Weissfeld L, Gandley RE, Roberts JM. Small low-density lipoproteins and vascular cell adhesion molecule-1 are increased in association with hyperlipidemia in preeclampsia. Metabolism 1998;47:1281-88.
18. Parra M, Rodrigo R, Barja P, Bosco C, et al. Screening test for preeclampsia through assessment of uteroplacental blood flow and biochemical markers of oxidative stress and endothelial dysfunction. Am J Obstet Gynecol 2005;193:1486-91.
19. Holthe M, Staff A, Berge L, Lyberg T. Different levels of platelet activation in preeclamptic, normotensive pregnant, and nonpregnant women. Am J Obstet Gynecol 2004;190:1128-34.
20. Redman C, Sacks G, Sargent I. Preeclampsia: An excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol 1999;180:499-506.
21. Redman C, Sargent I. Preeclampsia, the placenta and the maternal systemic inflammatory response, a review. Placenta 2003;24:s21-s27.
22. Benyo D, Miles T, Conrad K. Hypoxia stimulates cytokine production by villous explants from the human placenta. J Clin Endoc Metab 1997;82:1582-8.
23. Greer I, Lyall F, Perera T, Boswell F, et al. Increased concentrations of cytokines interleukin-6 and interleukin-1 receptor antagonist in plasma of women with preeclampsia, a mechanism for endotelial dysfunction? Obstet Gynecol 1994;84:937-40.
24. Redman C, Sargent I. Placental debris, oxidative stress and preeclampsia. Placenta 2000;21:597-602.
25. Young B, Levine R, Karumanchi A. Pathogenesis of preeclampsia. Annu Rev Pathol Mech Dis 2010;5:173-92.
26. Roberts J, Hube C. Is oxidative stress the link in the two-stage model of pre-eclampsia? Lancet 1999;354:788-89.
27. Gupta S, Agarwal A, Sharma R. The rol of placental oxidative stress and lipid peroxidation in preeclampsia. Obstet Gynecol Surv 2005;60:807-16.
28. Raijmakers M, Dechend R, Poston L. Oxidative stress and preeclampsia. rationale for antioxidant clinical trials. Hypertension 2004;44:374-380.
29. Dechend R, Viedt C, Muller DN, Ugele B, et al. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation 2003;107:1632-9.
30. Rodrigo R, Parra M, Bosco C, Fernández V, et al. Pathophysiological basis for the prophylaxis of preeclampsia through early supplementation with antioxidant vitamins. Pharmacol Therap 2005;107:177-97.
31. Atamer Y, Kocyigit Y, Yokus B, et al. Lipid peroxidation, antioxidante defense, status of trace metals and leptin levels in preeclampsia. Eur J Obstet Gynecol Reprod Biol 2005;119:60-6.
32. Staff A, Halvorsen B, Ranheim T, Henriksen T. Elevated level of free 8-iso-prostaglandin F2a in the decidua basalis of women with preeclampsia. Am J Obstet Gynecol 1999;181:1211-5.
33. Davidge ST, Signorella AP, Hubel CA, et al. Distinct factors in plasma of preeclamptic women increase endothelial nitric oxide or prostacyclin. Hypertension 1996;28:758-64.
34. Morris JM, Gopaul NK, Endresen MJ, Knight M, et al. Circulating markers of oxidative stress are raised in normal pregnancy and pre-eclampsia. Br J Obstet Gynecol 1998;105:1195-99.
35. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972;26:239-57.
36. Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 1995;146:3-15.
37. Huppertz B, Herrler A. Regulation of proliferation and apoptosis during development of the preimplantattion embryo and the placenta. Birth Def Res (part C) 2005;75:249-61.
38. Huppertz B, Kingdom JC. Apoptosis in the trophoblastdrole of apoptosis in placental morphogenesis. J Soc Gynecol Investig 2004;11:353-62.
39. Lunetta P, Penttila A. Immunohistochemical identification of syncytiotrophoblastic cells and megakaryocytes in pulmonary vessels in a fatal case of amniotic fluid embolism. Int J Legal Med 1996;108:210-4.
40. Huppertz B, Kadyrov M, Kigdom JC. Apoptosis and its role in the trophoblast. Am J Obstet Gynecol 2006;195:29-39.
41. Knight M, Redman CW, Linton EA, Sargent IL. Shedding of syncytiotrophoblast microvilli into the maternal circulation in pre-eclamptic pregnancies. Br J Obstet Gynaecol 1998;105(6):632-40.
42. Johansen M, Redman CW, Wilkins T, Sargent IL. Trophoblast deportation in human pregnancy—its relevance for pre-eclampsia. Placenta1999;20:531-9.
43. Formigli L, Papucci L, Tani A, Schiavone N, Tempestini A, Orlandini GE, et al. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J Cell Physiol 2000;182:41-9.
44. Levy R, Smith SD, Chandler K, Sadovsky Y, Nelson DM. Apoptosis in human cultured trophoblasts is enhanced by hypoxia and diminished by epidermal growth factor. Am J Physiol Cell Physiol 2000;278:C982-C988.
45. Tjoa ML, Cindrova-Davies T, Spasic-Boskovic O, Bianchi DW, Burton GJ. Trophoblastic oxidative stress and the release of cell-free feto-placental DNA. Am J Pathol 2006;169:400-4.
46. Straszewski S, Abrahams V, Mor G. The role of apoptosis in the regulation of trophoblast survival and differentiation during pregnancy. Endocrine Rev 2005;26:877-97.
47. Gupta AK, Rusterholz C, Huppertz B, Malek A, Schneider H, Holzgreve W, et al. A comparative study of the effect of three different syncytiotrophoblast microparticles preparations on endothelial cells. Placenta 2005;26:59-66.
48. Cockell AP, Learmont JG, Smarason AK, Redman CW, Sargent IL, Poston L. Human placental syncytiotrophoblast microvillous membranes impair maternal vascular endothelial function. Br J Obstet Gynaecol 1997;104:235-40.
49. Hoegh AM, Tannetta D, Sargent I, Borup R, Nielsen FC, Redman C, et al. Effect of syncytiotrophoblast microvillous membrane treatment on gene expression in human umbilical vein endothelial cells. BJOG 2006;113:1270-9.
50. Levine R, Karumanchi A. Circulating angiogenic factors in preeclampsia. Clin Obstet Gynecol 2005;48:372-86.
51. Kendall RL, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci USA 1993;90:10705-9.
52. He Y, Smith SK, Day KA. Alternative splicing of vasculal endothelial growth factor (CEGF)-R1 (FLT-1) pre m-RNA is important for the regulation of VEGF activity. Mol Endocrinol 1999;13:537-45.
53. Maynard S, Epstein F, Karumanchi A. Preeclampsia and angiogenic imbalance. Annu Rev Med 2008;59:61-78.
54. Levine R, Maynard S, Qian C, Lim KH, et al. Circulating angiogenic factors and the risk of preeclampsia. N Eng J Med 2004;350:672-83.
55. Chaiworapongsa T, Romero R, Espinoza J, et al. Evidence supporting a role for blockade of the vascular endothelial growth factor system in the pathophysiology of preeclampsia. Young Investigator Award. Am J Obstet Gynecol 2004;190:1547-50.
56. Maynard SE, Min JY, Merchan J, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 2003;111:649-58.
57. Ahmad S, Ahmed A. Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia. Circ Res 2004; 95: 884-91.
58. Li Z, Zhang Y, Ma J, Kapoun A, et al. Recombinant vascular endothelial growth factor 121 attenuates hypertension and improves kidney damage in a rat model of preeclampsia. Hypertension 2007;50:686-92.
59. Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endotelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427-34.
60. Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem 1999;274:584-94.
61. Toporsian M, Gros R, Kabir MG, et al. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ Res 2005;96:684-92.
62. Levine RJ, Lam C, Qian C, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med 2006;355:992-1005.
63. Venkatesha S, Toporsian M, Lam C, Hanai J. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med 2006;12:642-49.
64. Wang A, Rana S, Karumanchi A. Preeclampsia: The role of angiogenic factors in its pathogenesis. Physiology 2008;24:147-158.
65. Redman C, Sargent I. Latest advances in understanding preeclampsia. Science 2005;308:1592-94.
66. James J, Whitley G, Cartwright J. Preeclampsia: fitting together the placental, immune and cardiovascular pieces. J Pathol 2010;221:363-78.
67. Borzychowski A, Sargent I, Redman C. Inflammation and pre-eclampsia. Sem Fetal Neonatal Med 2006;11:309-316.
68. Redman C, Sargent I. Placental stress and Ppreeclampsia: A revised view. Placenta 2009;23:s38-s42.
69. Vatten L, Skjaerven R. Is preeclampsia more than one disease? Br J Obstet Gynecol 2004;111:298-302.
70. Roberts JM, Catov J. Preeclampsia more than 1 disease: or is it? Hypertension 2008;51:989-90.
71. Banias BB, Devoe LD, Nolan TE. Severe preeclampsia in preterm pregnancy between 26 and 32 weeks’ gestation. Am J Perinatol 1992;9:357-60.
72. Funai E, Friedlander Y, Palitel O, Tiram E, et al. Long-term mortality after preeclampsia. Epidemiology 2005;16: 206-15.
73. Irgens H, Reisaeter L, Irgens L, Lie R. Long term mortality of mothers and fathers after preeclampsia: population based cohort study. BMJ 2001;323:1213-17.
74. Sibai B. Diagnosis and Management of gestational hypertension and preeclampsia. Obstet Gynecol 2003;102:181-92.
75. Hauth JC, Ewell MG, Levine RL, Esterlitz JR, Sibai BM, et al. Pregnancy outcomes in healthy nulliparas women who subsequently developed hypertension. Obstet Gynecol 2000;95:24-8.
76. Hnat MD, Sibai BM, Caritis S, Hauth J, et al. Perinatal outcome in women with recurrent preeclampsia compared with women who develop preeclampsia as nulliparas. Am J Obstet Gynecol 2002;186:422-6.


