2020
6
Autores:
Sochog
Instituciones:
Wilmar Saldarriaga-Gil1, Estefanía Tascon Ospina2, Enrique Herrera-Castañeda3.
1 Especialista en ginecología y obstetricia, Magíster en ciencias básicas medicas, Doctor en genética medica. Profesor Titular Universidad del Valle. Ginecólogo Obstetra en Hospital Universitario del Valle. Cali, Colombia.
2 Medico en especialización en Ginecología y obstetricia,Universidad del Valle. Cali, Colombia.
3 Especialista en ginecología-obstetricia y en Medicina Reproductiva. Profesor Titular Universidad del Valle. Unidad de Medicina Reproductiva en Centro Médico Imbanaco. Cali, Colombia.
Correspondencia: Wilmar Saldarriaga Gil, wilmar.saldarriaga@correounivalle.edu.co
Tipo de documento:
Casos ClínicosSíndrome del X frágil en fecundación in vitro. Reporte de caso
Contenido del documento:
INTRODUCCIÓN
El síndrome X frágil (SXF), OMIM # 300624, es un trastorno genético heredado a través del cromosoma X. Es clasificado entre las enfermedades por repetición de tripletas; es la segunda causa de discapacidad intelectual (DI) de origen genético después del síndrome de Down y es la primera causa heredable de DI y autismo. El fenotipo clásico también incluye cara alargada, orejas grandes y aladas, hiperextensibilidad y macroorquidismo. En mujeres el fenotipo es menos especifico, esto secundario a la variabilidad de la inactivación de cromosoma X con la alteración genética. El 60% tienen déficit cognitivo pero de menor severidad que en hombres1.
El SXF se produce por la ausencia de la FMRP (sigla del inglés Fragile X Mental Retardation Protein), particularmente en cerebro en donde su función es inhibir la traducción de mRNAs neuronales que codifican para proteínas implicadas en la en la conectividad sináptica, y en tejido conectivo donde hace parte de la cascada de proteínas que producen la remodelación de la matrix extracelular; esta proteína es codificada por el gen FMR1 (Fragile X Mental Retardation 1), locus Xq27,3, el cual tiene, en su región corriente arriba no codificante, un número variable de tripletas CGG. El número de repeticiones de la tripleta produce cuatro variantes alélicas: normal con 44 o menos repeticiones, zona gris entre 45 y 54 repeticiones, premutación (PM) entre 55 y 200, y mutación completa si hay más de 200 repeticiones. La mutación completa es la que se asocia al fenotipo SXF, en diferentes grados de expresión1.
Las mujeres portadoras de la PM tienen varias implicaciones clínicas, como la posible expansión del número de tripletas a sus descendientes; el riesgo de expansión aumenta a mayor número de repeticiones de la tripleta CGG en el gen FMR1 de la portadora. En cada embarazo, las portadoras de la PM tienen un riesgo del 50% de transmitir el alelo anormal a sus descendientes quienes serían afectados por el SXF2. Adicionalmente, se han descrito tres subfenotipos relacionados con el SXF en portadoras de la premutación. FXPOI (del inglés Fragile X premature ovarian insufficiency), caracterizado por ausencia de menstruación antes de los 40 años y disminución de la reproducción; FXTAS (del inglés Fragile X Tremor Ataxia Associated Syndrome), en el que se presenta temblor de intención y ataxia después de los 50 años, y por último, FXAND (del inglés Fragile X neuropsychiatric disorders), caracterizado por ansiedad, depresión, y neuropatías periféricas3.
La reproducción asistida facilita o sustituye procesos naturales de fecundación e implantación del embrión y con frecuencia requiere de donantes de óvulos o semen para lograr el embarazo. Los procesos de selección de donantes de gametos incluyen estudio de la historia médica, laboratorios para enfermedades infecciosas y cariotipo, entre otros4. Múltiples instituciones de reproducción asistida han adicionado a sus protocolos de evaluación de donantes, pruebas moleculares para identificar variantes genéticas asociadas a enfermedades de herencia mendeliana recesiva, estos análisis usualmente incluyen el gen FMR15,6.
El objetivo de este reporte es presentar un caso de SXF producto de reproducción asistida, y proponer que en Colombia se actualice la normatividad que rige las pruebas aplicadas a posibles donantes de gametos, y parejas o individuos en protocolos de reproducción asistida, incluyendo pruebas que identifiquen portadores asintomáticos de enfermedades genéticas como el SXF.
PRESENTACIÓN DEL CASO
Una pareja consultó a unidad de medicina reproductiva; la mujer, de 30 años, recibía terapia de reemplazo hormonal para menopausia precoz secundaria a hipofisectomía indicada por un hiperprolactinoma. El hombre, de 36 años, no tenía enfermedades crónicas ni antecedentes familiares relevantes, y presentó un espermograma sin alteraciones.
La pareja se sometió a un ciclo de estimulación con HMG, sin respuesta terapéutica. En consenso, el grupo médico y la pareja deciden realizar fecundación in vitro (FIV), con protocolo con óvulo donado.
Donante anónima de óvulos: Mujer de 24 años, sana, a quien se le aplicaron los protocolos de donante de óvulos recomendados por el Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA) de Colombia, para las unidades de medicina reproductiva. La donante negó antecedentes de enfermedades crónicas relevantes, anomalías congénitas y enfermedades genéticas en ella o en su familia; también negó antecedentes familiares de DI, autismo y enfermedades psiquiátricas. Tampoco manifestó exposición a teratógenos como medicamentos, psicoactivos, o elementos químicos o físicos como radiación, entre otros. Historia reproductiva: G0P0, sin donación de óvulos previa. Al examen físico: etnia latina, IMC 22, sin otros hallazgos particulares. Las siguientes pruebas fueron negativas: Virus de la inmunodeficiencia humana (VIH), Antígeno de superficie para hepatitis B, Inmunoglobulina G y M anti Core, Inmunoglobulina G para hepatitis B, serología para sífilis RPR, cultivo para Neisseria gonorrhoeae, Inmunoglobulina G anti Citomegalovirus y fracción Beta de la hormona gonadotropina coriónica humana; el hemograma y los tiempos de coagulación estuvieron dentro de límites normales. El resultado del cariotipo de bandeo G fue 46,XX, en 30 metafases, interpretado como ausencia de alteraciones cromosómicas numéricas o estructurales identificables por dicha técnica.
A la donante se le realizó protocolo de inducción de ovulación comenzando con acetato de leuprolide 2 cm3 subcutaneos en la fase lutea del ciclo anterior, se inició estimulación el día 3 del ciclo con 75 unidades internacionales (UI) de FSH y 75 UI de LH, se realizo estimulación con citrato de clomifeno por 11 dias, y seguimiento de ecográfico folicular y de estradios interdiario; se evidenciaron 9 foliculos en ovario izquierdo y 11 en el derecho, cuando se observaron 3 foliculos de mas de 18 mm, se aplicaron 5000 UI de Hormona Gonadotropina Corinica y 36 horas posterior a esa aplicación se le realizó aspiración folicular, obteniendo 8 óvulos; estos se llevaron a fertilización in vitro, en la cual se produjo el desarrollo adecuado de 4 embriones, 2 de los cuales fueron transferidos.
El protocolo logró un embarazo gemelar bicorial biamniótico, seguido mediante 10 visitas de control prenatal y 6 ecografías, entre ellas una de detalle anatómico; ninguna ecografía detectó anomalías congénitas ni alteraciones del crecimiento fetal. La gestación se finalizó por cesárea a las 35 semanas y 6 días por un trastorno hipertensivo del embarazo severo. Se obtuvo un primer recién nacido (gemelo 1), femenino, en presentación podálica, con los siguientes parámetros: peso 2500 gramos, talla 46 cm, perímetro cefálico 34 cm, Apgar de 9 y 10 (1´y 5´, respectivamente). El segundo recién nacido (gemelo 2), masculino, tuvo los siguientes parámetros: peso 2100 gr, talla 45 cm, PC 33 cm, Apgar de 9 y 10 (1´y 5´, respectivamente). El gemelo 2, en seguimiento hasta los 5 años, tiene un desarrollo psicomotor adecuado.
La gemela 1 fue valorada a los 19 meses de edad por pediatra infectólogo, debido a un cuadro de bronconeumonía de difícil manejo; en esta consulta se detectó un retraso global del desarrollo: inició gateo a los 12 meses, aún no caminaba, solo pronunciaba monosílabos, se mostraba ansiosa e irritable y temorosa ante extraños. En el examen físico se observó hoyuelo en la porción anterior y superior de la oreja derecha y estrabismo en ojo izquierdo. Dados los hallazgos fenotípicos se consideró sospecha de SXF y se realizo PCR con doble cebador para cuantificación de tripletas del gen FMR1, que identificó un alelo con 29 repeticiones de la tripleta CGG y el otro con más de 200 repeticiones, resultado que fue confirmado mediante Southern Blot. Se hizo diagnóstico de mutación completa en el gen FMR1 y de SXF.
Con los anteriores hallazgos, se contactó a la donante de óvulos, quien aceptó realizarse la misma prueba molecular. En la donante, el PCR con doble cebador mostró un alelo del gen FMR1 con 30 repeticiones de la tripleta CGG y el otro con más de 68 repeticiones, resultado confirmado también por Southern blot. El diagnóstico de la donante fue portadora de la premutación del FMR1; recibió asesoría genética y reproductiva, sobre la probabilidad de tener hijos con la PM, la MC y SXF, así como de desarollar FXTAS y FXPOI.
La gemela 1 diagnosticada con SXF, ahora con 5 años, recibe terapia física, ocupacional y del lenguaje, con buena respuesta. A los 4 años y 10 meses tuvo cirugía para corrección de estrabismo. Al momento de este reporte, asiste al jardín de infantes y alcanza, aunque con mayor dificultad que sus compañeros, las metas escolares, y ha logrado una integración parcial a las actividades de clase.
DISCUSIÓN
La prevalencia en mujeres de la premutación del gen FMR1 relacionada con el SXF en la población general es variable, y depende del número de tripletas CGG en el gen FMR1 utilizadas para el diagnóstico y de la población estudiada. En Finlandia, Ryyna ̈nen et al, realizaron un estudio prospectivo con 1,477 pacientes embarazadas y encontró una prevalencia de portadoras de la PM de 1 en 246, utilizando como criterio alelos con más de 60 repeticiones7 . En Israel, Pesso et al, reportó que 1 de cada 70 mujeres tenía 52 o más repeticiones, y 1 de cada 152 pacientes tenía más de 54 repeticiones de CGG8. En Israel, Hagit et al, en un estudio prospectivo, donde se incluyeron 14,334 mujeres embarazadas o en estado preconcepcional, sin historia familiar o personal de discapacidad intelectual, se les realizó Southern blot para determinar la presencia de expansiones en el gen FMR1, y si la prueba era positiva se procedía a PCR para determinar el número de tripletas CGG. En este estudio, 1 de cada 113 mujeres tenía la PM en FMR1 9 . Tassone et al, en Estados Unidos de America, a través de un estudio de tamizaje en 14,207 recién nacidos, reportó una prevalencia de alelos con la PM de 1 en cada 209 recién nacidos femeninos y de 1 en cada 430 recién nacidos masculinos10. En la literatura se acepta que la prevalencia de PM es de 1 en cada 200 a 300 mujeres11.
En Colombia no encontramos estudios sobre la prevalencia de la PM del gen FMR1 en población general. Sin embargo, en Ricaurte, un corregimiento colombiano ubicado en el departamento del Valle del Cauca, y que es considerado un conglomerado genético de SXF, se describieron las prevalencias más elevadas de los alelos de la PM y MC del FMR1 descritas en la literatura. En Ricaurte se identificaron 20 portadoras con tripletas entre 55 y 200 repeticiones entre 502 mujeres evaluadas, mostrando que 1 de cada 25 mujeres es portadora de la PM; esta prevalencia es 11,1 veces más elevada que las prevalencias globalmente reportadas 12. No existen reportes que establezcan o estimen la prevalencia o número de casos de PM para el resto del país12.
Las mujeres con la PM del gen FMR1 tienen trastornos reproductivos y endocrinológicos; las portadoras de la PM tienen 20 veces más probabilidad de presentar menopausia precoz (ausencia de menstruación antes de los 40 años). Allingham-Hawkins DJ et al, en 1999, encontró, entre 395 mujeres con la PM, una prevalencia de menopausia precoz del 16%, mientras que en la población general se ha reportado en alrededor del 1% 13. También se ha reportado la menopausia, aún después de los 40 años, es más temprana. Adicionalmente, la insuficiencia ovárica en mujeres con la PM, entre los 18 y 30 años esta entre 1,4% y 3%, mientras que en la población general, en el mismo rango de edad, es de 0,1% 14. Dado lo anterior, se debe realizar PCR para cuantificar tripletas del gen FMR1 en mujeres con falla ovárica prematura. La donante de óvulos portadora de la PM no tenía signos de insuficiencia ovárica.
Las mujeres candidatas a donante de óvulos o que asisten a consulta preconcepcional, pueden tener un examen médico sin hallazgos sugestivos, y por tanto, se les debería practicar pruebas para cuantificar el número de tripletas CGG del FMR1 8. En Colombia las recomendaciones del INVIMA para donantes de gametos, ordenan realizar cariotipo con bandas G, el cual solo puede diagnosticar alteraciones cromosómicas numéricas y estructurales, pero no incluyen pruebas moleculares para evaluar el número de tripletas del FMR1, ni tampoco para detectar alteraciones en otros genes implicados en enfermedades genéticas de herencia recesiva 4.
La PCR con doble o triple cebador y el Southern Blot (SB) son las pruebas moleculares de tamizaje y confirmación, respectivamente, de las variantes alélicas PM y MC del gen FMR1. Estas pruebas se pueden aplicar a diferentes muestras; en diagnóstico prenatal, en DNA extraído por biopsia de vellosidad corial, en líquido amniótico o en sangre fetal, y en niños o adultos en muestras de sangre periférica. La PCR y el SB pueden identificar la PM y la MC y cuantificar las tripletas; además el SB puede determinar el estado de metilación de las islas CpG del gen FMR1. En comparación con la PCR, el SB requiere más DNA, toma más tiempo, y es más costoso, por tanto se utiliza solo como prueba confirmatoria de las variantes alélicas relacionadas con el SXF15. Al caso aquí reportado, a su gemelo y a la donante de óvulos, se les realizó tanto PCR como SB.
En el diagnóstico genético preimplantación (DP), la PCR es la prueba molecular indicada para la cuantificación de tripletas. A través de biopsia de blastómera o de trofoectodermo de blastocisto, se puede obtener suficiente DNA para realizar una PCR con doble o triple cebador. Sin embargo, algunos autores sugieren que el PCR no es concluyente y proponen que se complemente con análisis de haplotipos flanqueantes al FMR1 con técnicas de secuenciación de siguiente generación. En el DGP el SB no puede utilizarse dado que las cantidades DNA obtenidas no son suficientes para realizar la prueba. Así es que, las pruebas moleculares para el SXF en diagnóstico preimplantación son posibles y se realizan en laboratorios especializados16–18.
La asesoría genética y reproductiva en portadoras de la PM del FMR1 debe incluir varios tópicos. El primero, es explicar la probabilidad, en términos de porcentaje, de tener hijos con el alelo normal, con la PM o con la MC y de ser afectados por el SXF. El segundo, tratar las opciones de diagnóstico prenatal en embarazos espontáneos, como la prueba de cuantificación de tripletas en DNA extraído por biopsia de vellosidad corial, líquido amniótico o sangre fetal. Tercero, exponer las opciones de diagnóstico preimplantacional en casos de reproducción asistida. El cuarto es sugerir pruebas moleculares a otros integrantes de la familia que podrían tener PM o MC. Finalmente, se deben explicar los síntomas de FXTAS, FXPOI y FXAND, y las probabilidades de padecerlos (ver tabla1). Se sugiere entregar un informe escrito que describa estos tópicos, para que el paciente, su familia y los profesionales sanitarios entiendan la trascendencia del resultado y, así, poder generar una toma de decisiones informada. En el caso aquí reportado, a la donante de óvulos con PM se le brindó asesoría genética y reproductiva.
En donantes de óvulos o semen se deben realizar pruebas moleculares para evaluar si sus gametos pudieran tener variantes alélicas causantes de enfermedades genéticas en los embarazos generados a partir de ellos. Las Sociedades Americana y Europea de Medicina Reproductiva recomiendan pruebas moleculares a los donantes de gametos si hay hallazgos en la historia familiar relacionados con enfermedades de origen genético, o si pertenecen a un grupo étnico reconocido por presentar enfermedades genéticas con mayor frecuencia que en la población general. En Estados Unidos de América se realizan pruebas para fibrosis quística y atrofia muscular espinal, no solo a donantes de gametos, sino a toda la población en edad reproductiva 5,19–21.
Basados en estas recomendaciones, la población general oriunda de Ricaurte o cuyos sus padres son originarios de Ricaurte, especialmente si son candidatos a donantes de gametos, deben realizarse PCR para evaluar si tienen variantes alélicas del FMR1 relacionadas con el SXF.
Con la tecnología de la secuenciación de siguiente generación se tienen nuevas posibilidades para evaluar a parejas, tanto en cita preconcepcional como en reproducción asistida, y a donantes de gametos. El Instituto Valenciano de Infertilidad (IVI) y otros centros de reproducción asistida, en sus protocolos recomiendan realizar paneles moleculares que evalúan alteraciones en al menos 600 genes, relacionados con enfermedades monogénicas y herencia recesiva como fibrosis quística, hemoglobinopatías, errores congénitos del metabolismo y SXF. Específicamente, en los casos en que los donantes tengan una o algunas variantes en heterocigosidad en genes relacionados con patologías genéticas, se sugieren dos opciones a seguir: la primera es descartar el donante, y la segunda, es realizar una evaluación dirigida a ese gen (por ejemplo, mediante secuenciación sanger) en el receptor; si el receptor tiene alguna variante genética implicada en la misma patología, se debe descartar al donante, pero si no la tuviera, se asesoraría a la pareja receptora explicando que el recién nacido producto de ese gameto tendría 50 % de probabilidad de ser portador de esa variante, y una probabilidad cercana a 0 (cero) de padecer la enfermedad genética. Con la información, será la pareja receptora quien tome la decisión de continuar el proceso de FIV con ese donante y realizar diagnóstico genético preimplantacional específico para el alelo implicado. La aplicación de pruebas moleculares a donantes de gametos puede disminuir la incidencia de casos de enfermedades con origen genético en reproducción asistida5.
El caso aquí reportado, de una donante de óvulos a quien se le realizó el protocolo con las pruebas ordenadas por el INVIMA, sin antecedentes familiares de discapacidad intelectual y autismo y sin hallazgos sospechosos de ser portadora de la PM del FMR1, y que generó como producto un recién nacido con SXF, aporta argumentación para sugerir que la normatividad y los protocolos de las clínicas de reproducción sean actualizados para incluir pruebas de detección de variantes genéticas implicadas en enfermedades monogénicas recesivas y SXF en donantes de óvulos y semen.
REFERENCIAS
- Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB, Moine H, Kooy RF, et al. Fragile X syndrome [Internet]. Vol. 3, Nature reviews. Disease primers. 2017 [cited 2020 Mar 30]. p. 17065. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28960184
- Saldarriaga W, Tassone F, González-Teshima LY, Forero-Forero JV, Ayala-Zapata S, Hagerman R. Fragile X syndrome. Colomb Med. 2014;45(4):190–8.
- Salcedo-Arellano MJ, Dufour B, McLennan Y, Martinez-Cerdeno V, Hagerman R. Fragile X syndrome and associated disorders: Clinical aspects and pathology. Vol. 136, Neurobiology of Disease. Academic Press Inc.; 2020.
- Decreto 1546 de 1998 – EVA – Función Pública [Internet]. [cited 2020 Mar 30]. Available from: https://www.funcionpublica.gov.co/eva/gestornormativo/norma.php?i=14522
- Dondorp W, De Wert G, Pennings G, Shenfield F, Devroey P, Tarlatzis B, et al. ESHRE Task Force on Ethics and Law 21: genetic screening of gamete donors: ethical issues. Hum Reprod [Internet]. 2014 Jul [cited 2020 Mar 30];29(7):1353–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24859980
- Practice Committee of American Society for Reproductive Medicine, Practice Committee of Society for Assisted Reproductive Technology. Recommendations for gamete and embryo donation: A committee opinion. Fertil Steril [Internet]. 2013 Jan [cited 2020 Mar 30];99(1):47–62. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23095142
- Ryynänen M, Heinonen S, Makkonen M, Kajanoja E, Mannermaa A, Pertti K. Feasibility and acceptance of screening for fragile X mutations in low-risk pregnancies. Eur J Hum Genet [Internet]. 1999 [cited 2020 Mar 30];7(2):212–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10196705
- Pesso R, Berkenstadt M, Cuckle H, Gak E, Peleg L, Frydman M, et al. Screening for fragile X syndrome in women of reproductive age. Prenat Diagn [Internet]. 2000 Aug [cited 2016 Aug 21];20(8):611–4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10951469
- Toledano-Alhadef H, Basel-Vanagaite L, Magal N, Davidov B, Ehrlich S, Drasinover V, et al. Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am J Hum Genet. 2001;69(2):351–60.
- Tassone F, Iong KP, Tong T-H, Lo J, Gane LW, Berry-Kravis E, et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med [Internet]. 2012 [cited 2016 Aug 21];4(12):100. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23259642
- Hunter J, Rivero-arias O, Angelov A, Kim E, Fotheringham I, Leal J, et al. Epidemiology of Fragile X Syndrome : A Systematic Review and Meta-Analysis. 2014;1648–58.
- Saldarriaga W, Forero-Forero JV, González-Teshima LY, Fandiño-Losada A, Isaza C, Tovar-Cuevas JR, et al. Genetic cluster of fragile X syndrome in a Colombian district. J Hum Genet [Internet]. 2018;63(4):509–16. Available from: https://doi.org/10.1038/s10038-017-0407-6
- Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, Holden JJA, Yang KT, Lee C, et al. Fragile X premutation is a significant risk factor for premature ovarian failure: The international collaborative POF in fragile X study – Preliminary data. Am J Med Genet. 1999 Apr 2;83(4):322–5.
- Gómez Tabares G, Saldarriaga W HL. Insuficiencia ovárica prematura. Rev colomb menopaus. 2018;8–18.
- Tassone F. Advanced technologies for the molecular diagnosis of fragile X syndrome. Expert Rev Mol Diagn [Internet]. 2015 [cited 2017 Jan 22];15(11):1465–73. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26489042
- Kieffer E, Nicod JC, Gardes N, Kastner C, Becker N, Celebi C, et al. Improving preimplantation genetic diagnosis for Fragile X syndrome: Two new powerful single-round multiplex indirect and direct tests. Eur J Hum Genet. 2016 Feb 1;24(2):221–7.
- Reches A, Malcov M, Ben-Yosef D, Azem F, Amit A, Yaron Y. Preimplantation genetic diagnosis for fragile X syndrome: Is there increased transmission of abnormal FMR1 alleles among female heterozygotes? In: Prenatal Diagnosis [Internet]. Prenat Diagn; 2009 [cited 2020 Sep 19]. p. 57–61. Available from: https://pubmed.ncbi.nlm.nih.gov/19097038/
- Rajan-Babu IS, Lian M, Cheah FSH, Chen M, Tan ASC, Prasath EB, et al. FMR1 CGG repeat expansion mutation detection and linked haplotype analysis for reliable and accurate preimplantation genetic diagnosis of fragile X syndrome. Expert Rev Mol Med. 2017 Jul 19;19.
- Committee opinion No. 486: Update on carrier screening for cystic fibrosis [Internet]. Vol. 117, Obstetrics and Gynecology. 2011 [cited 2020 Mar 30]. p. 1028–31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21422883
- Prior TW. Carrier screening for spinal muscular atrophy. Vol. 10, Genetics in Medicine. Various; 2008. p. 840–2.
- Reproducción asistida – Tratamientos de fertilidad – IVI [Internet]. 2019 [cited 2020 Sep 19]. Available from: https://ivi.es/
FIGURA
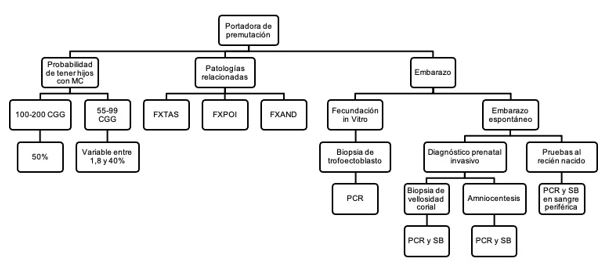
Figura 1. Asesoria genetica y reproductiva en portadora de la premutacion del gen FMR1 relacionado con el SXF. Se muestra como la asesoria debe incluir la probablidad e tener hijos con SXF, la explicacion de padecer patologias relacionadas como FXTAS, FXPOI, FXAND y las opciones de diangostico preinplantacion, prenatal y postnatal.


