2012
4
Autores:
Wilmar Saldarriaga1, Albaro José Nieto Calvache1, Juan Carlos Quintero2, Harry Pachajoa3, Carolina Isaza1
Instituciones:
1Universidad del Valle, 2Hospital Universitario del Valle, Centro Médico Imbanaco, 3Universidad Icesi. Cali, Colombia.
Imágen 1:
Imágen 2:
Imágen 3:
Tipo de documento:
Casos ClínicosSÍNDROME OTOPALATODIGITAL TIPO II, APROXIMACIÓN PRENATAL Y DIAGNÓSTICO CLÍNICO DE UN CASO COMPLEJO DE DISPLASIA ÓSEA
Contenido del documento:
RESUMEN
El síndrome otopalatodigital tipo 2 (OPD2), es una rara entidad con herencia recesiva ligada al cromosoma X, letal, caracterizada por facies anormales con hipoplasia centrofacial, hipertelorismo ocular, paladar hendido, talla baja, huesos largos curvos, sindactilia en pies y manos y anomalías óseas. Usualmente originadas en mutaciones en el gen de la filamina A (FLNA). Se reporta un caso, con diagnóstico prenatal de osteocondrodisplasia que posteriormente por hallazgos al examen físico y radiológicos del recién nacido se clasifico como síndrome otopalatodigital tipo 2.
PALABRAS CLAVE: Genética, osteocondrodisplasias, diagnóstico prenatal, anomalías músculo esqueléticas
SUMMARY
Otopalatodigital syndrome, type 2 (OPD2), is a rare entity with recessive heredity linked to the X chromosome, lethal, characterized by abnormal facies, with centro-facial hypoplasia, ocular hypertelorism, cleft palate, low height, curved long bones, syndactyly, and osseous anomalies on feet and hands. It has been recently shown that patients with OPD2 with mutations in the filamin A gene (FLNA), which is also found altered in allelic entities like the OPD1 syndrome, the Melnik-Needles syndrome and frontometaphyseal dysplasia. Herein, we report a case with prenatal osteochondrodysplasia diagnosis that after physical and radiological exam of the newborn was classified as otopalatodigital syndrome type 2.
KEY WORDS: Genetics, osteochondrodysplasias, prenatal diagnosis, musculoskeletal abnormalities
INTRODUCCIÓN
El síndrome de otopalatodigital, tipo 2 (OMIM OPD2: # 304120) es una rara entidad letal, con herencia recesiva ligada al cromosoma X, caracterizada por facies anormales con hipoplasia facial central, hipertelorismo ocular, paladar hendido, talla baja, huesos largos curvos, sindactilia, y anomalías óseas en pies y manos. Otros hallazgos menos frecuentes abarcan onfalocele, defectos renales de las vías urinarias. El fallecimiento suele ser perinatal, algunos pacientes solo alcanzan pocos meses de vida (1,2,3). Recientemente se ha demostrado que los pacientes con OPD 2 tienen mutaciones en el gen A filamina (FLNA), que también se encuentra alterado en entidades alélicas como el síndrome de OPD1, el síndrome Melnik-Needles, y displasia frontometafisaria (2-5). Robertson en 2005 (5), propuso el término osteodisplasia fronto-oto-palato-digital para agrupar estas cuatro entidades que tiene características fenotípicas comunes y el mismo gen alterado.
Se presenta el caso de un recién nacido con diagnóstico prenatal de osteocondrodisplasia, que después de un examen físico y radiológico fue clasificado como síndrome otopalatodigital tipo II (ODP II).
CASO CLÍNICO
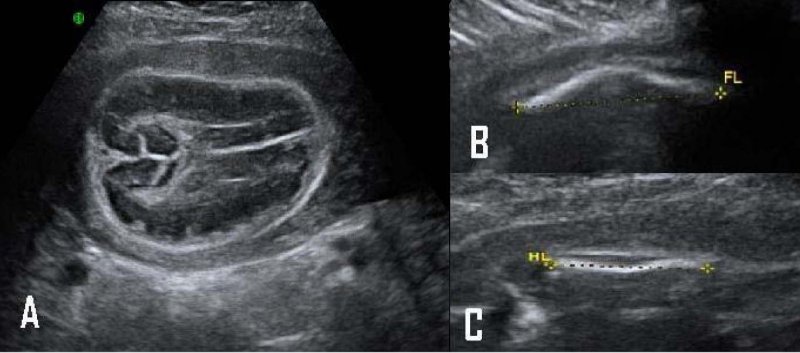
Recién nacido muerto de sexo indefinido, producto de embarazo de 28 semanas de edad gestacional, de padres consanguíneos, madre primigesta de 15 años de edad, sin antecedentes familiares de malformaciones óseas u otras anomalías congénitas. La madre asistió a cuatro controles prenatales y tres ecografías que se realizaron entre las semanas 26 y 28, describen un feto con alteraciones múltiples, anhidramnios, hipomineralización de los huesos del cráneo, acortamiento de huesos largos (marcador rizomélico, más grave en las extremidades superiores); además los huesos largos eran curvos con posibles fracturas, no se visualizaron manos ni pies (Figura 1). Por otra parte, los riñones se observaron hipoplásicos y alteración de la pared abdominal con un posible onfalocele. Con estos hallazgos, se realizó cordocentesis para cariotipo con bandas G, con resolución de 650 bandas, 46, XY, sin alteraciones estructurales.
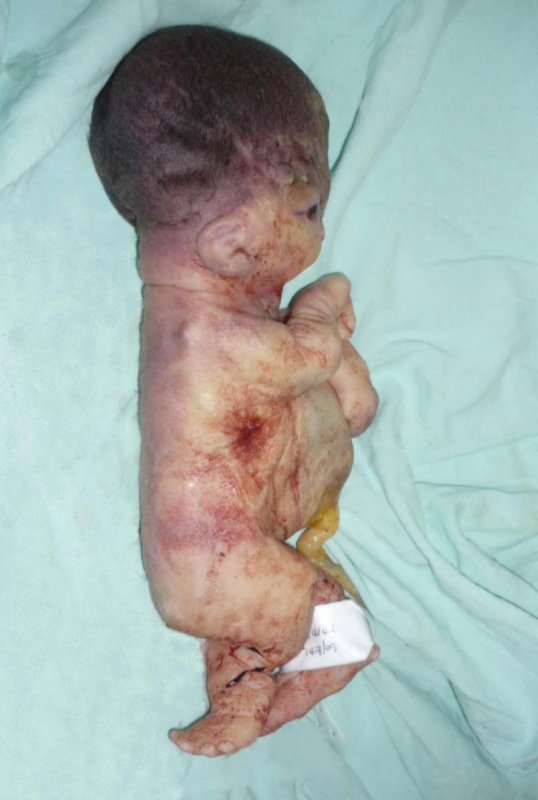
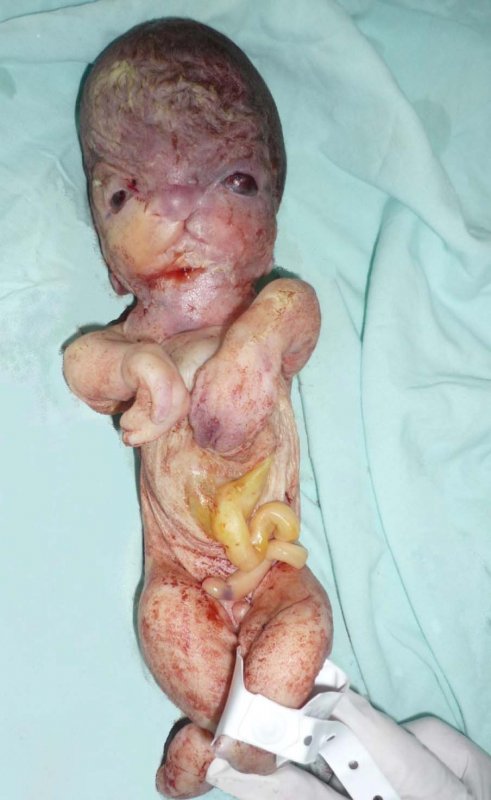
Por anomalías congénitas incompatibles con la vida, los padres solicitan interrupción voluntaria del embarazo. Por vía vaginal se obtiene un recién nacido muerto, de sexo indefinido, con un peso 745 gramos, menor de percentil 3, talla de 29 cm, menor de percentil 3. Microtia bilateral de tipo I, se observó agenesia del canal auditivo externo, hipertelorismo ocular severo, frente amplia, el labio fisurado central, paladar fisurado, boca pequeña, micrognatia severa, cuello corto, tórax estrecho, con abdomen deformado por protrusión de vísceras abdominales, pero contenidas por pared abdominal muy delgada, además inserción de cordón umbilical muy amplia, sin salida de órganos abdominales a través de la hernia umbilical (Figuras 2, 3, y 4). También se evidencia sindactilia completa de manos y pies. En los estudios radiológicos se observó hemivértebra cervical, hipoplasia severa de la mandíbula, los huesos largos curvos, adelgazados y angulados (Figura 5). La imagen de la tomografía axial revela la profusión de las vísceras abdominales (Figura 6).
El informe del caso y la publicación de las fotografías se realizan con el consentimiento informado firmado de los padres.
DISCUSIÓN
Las displasias óseas son un grupo de trastornos del desarrollo del esqueleto que son genéticamente y clínicamente heterogéneas. Su prevalencia estimada es de 1 por cada 4000 nacimientos, e incluye las displasias que se caracterizan por la osteogénesis imperfecta y las osteocondrodisplasias. Las osteocondrodisplasias son el resultado de mutaciones de los genes esenciales para el desarrollo de huesos y cartílagos. La presentación clínica está dominada por anomalías esqueléticas, sus manifestaciones pueden ser limitadas en el hueso, pero en la mayoría de los casos el tejido no esquelético esta alterado (6).
En las osteocondrodisplasias se produce talla baja, con desproporción entre los segmentos corporales. Se clasifican de acuerdo a las estructuras óseas comprometidas, como el cráneo, los huesos largos (epífisis, metafísis o diáfisis), vértebras y costillas. Por estas características la evaluación radiográfica de estos pacientes debe incluir imágenes completas de la totalidad del esqueleto (6).
Aunque algunas condrodisplasias pueden ser fácilmente diagnosticadas, la mayoría requieren de un análisis de la historia clínica que incluya información de la familia, un examen físico exhaustivo, radiografías, exámenes de laboratorio y cariotipo de alta resolución para descartar alteraciones cromosómicas numéricas o estructurales. El proceso implica el reconocimiento de patrones complejos de compromiso óseo y defectos congénitos asociados, estos últimos característicos de los diferentes síndromes con condrodisplasias, teniendo en cuenta que algunos de ellos son incompatibles con la vida.
Aquí, presentamos un caso con hallazgos en ecografía prenatal, examen físico y radiológicos del recién nacido con características faciales, óseas, mas sindactilia, defectos renales y de pared abdominal, que llevan al diagnostico clínico de síndrome de otopalatodigital tipo 2 (OPD2).
El síndrome OPD2 es causado por una mutación en el gen FNLA con locus Xq28, que codifica la proteina filamina A (5). Esta proteína pesa 280 KD y es una proteína de unión a actina, que entreteje los filamentos de actina en las redes ortogonales en el citoplasma cortical y participa en el anclaje de proteínas de membrana a través del citoesqueleto de actina. La remodelación del citoesqueleto es crucial para la modulación de la forma celular y la migración.
Filamina A es una proteína que regula la organización del citoesqueleto de actina mediante la interacción con integrinas, los complejos de receptores transmembrana y el sistema de segundos mensajeros (7,8). La pérdida de una función de la filamina por lo general produce un estado embrionario letal en los varones, y en las mujeres se manifiesta como un trastorno en la migración neuronal periventricular llamado heterotopia (8).
Patrones de herencia recesiva, dominante y semi-dominante se han descrito ligada al cromosoma X (1,2), con una probabilidad de repetición de 25 a 50%, por lo cual se requiere idealmente un estudio molecular del caso y de la madre.
Hay pocos casos de síndrome de OPD2 reportados en la literatura, probablemente debido a su alta tasa de mortalidad en útero o la temprana mortalidad en la vida postnatal, produciendo poco interés en los padres o en los médicos tratantes. Aquí presentamos un nuevo caso de OPD2 con un enfoque prenatal orientado en los hallazgos de la ecografía obstétrica hacia una displasia ósea letal, con compromiso renal y onfalocele. Los hallazgos del examen físico del recién nacido, las imágenes radiológicas, y un estudio cromosómico normal, permitieron llegar al diagnóstico clínico de síndrome oto-palato-digital tipo 2, con lo cual se logró proporcionar asesoramiento genético y reproductivo adecuado a una pareja con expectativa reproductiva, guiado por un grupo interdisciplinario de obstetras y genetistas.
CONCLUSIÓN
Se reporta un caso de síndrome de osteocondrodisplasia asociado a alteraciones craneofaciales, renales y de pared abdominal, clasificado como síndrome otopalatodigital tipo 2. El diagnóstico prenatal permitió al equipo multidisciplinario asesorar a la familia en la toma de decisiones.
BIBLIOGRAFÍA
1. Marino-Enriquez A, Lapunzina P, Robertson SP, Rodriguez JI. Otopalatodigital syndrome type 2 in two siblings with a novel filamin A 629G-T mutation: clinical, pathological, and molecular findings. Am J Med Genet 2007;143A:1120-5.
2. Mckusick V, Bocchini C, Kniffin C. Online Mendelian Inheritance in Man OMIM [base de datos en Internet]. OTOPALATODIGITAL SYNDROME, TYPE II; OPD2. Baltimore: Johns Hopkins University. [Actualizada 28 de Julio de 2009; acceso 10 abril 2009]. URL disponible en: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=304120
3. Young K, Barth CK, Moore C, Weaver DD. Otopalatodigital syndrome type II associated with omphalocele: report of three cases. Am J Med Genet 1993;45:481-7.
4. Verloes A, Lesenfants S, Barr M, Grange DK, Journel H, Lombet J, et al. Fronto-otopalatodigital osteodysplasia: clinical evidence for a single entity encompassing Melnick-Needles syndrome, otopalatodigital syndrome types 1 and 2, and frontometaphyseal dysplasia. Am J Med Genet 2000;90:407-22.
5. Robertson SP. Filamin A: phenotypic diversity. Curr Opinion Genet Dev 2005;15:301-7.
6. Firth H, Hurst A. Oxford desk reference: Clinical genetics. Oxford University Press. 2007.
7. Tu Y, Wu S, Shi X, Chen K, Wu C. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell 2003;113:37-47.
8. Robertson SP, Twigg SR, Sutherland-Smith AJ, Biancalana V, Gorlin RJ, Horn D, et al. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nature Genet 2003;33:487-91.
9. Johnson JN, Hartman TK, Krych EH, Seferian EG, Ouellette Y. Tracheomalacia in siblings with otopalatodigital syndrome. (Letter) Am J Med Genet 2008;146A:1347-9.
OBSERVACIONES
Agradecimientos: Damos las gracias a todos los colaboradores de vigilancia epidemiológica de malformaciones congénitas en el Hospital Universitario del Valle (HUV), que utiliza la metodología del ECLAMC. Agradecemos también al grupo de investigación de MACOS y el comité de medicina materno fetal en el HUV.


